Fundamental signaling pathways for glioblastoma drug resistance and developing robust organoid models for drug discovery
Article information

Abstract
This article presents a review of the current literature on the molecular mechanisms of treatment resistance in glioblastoma. As mounting research continues to explore novel methods of treating glioblastoma, from using organoid models for drug screening to developing novel cellular therapies, it is critical to understand the fundamental molecular landscape that makes glioblastoma difficult to treat. This review explores the means of chemoresistance to the conventional chemotherapy temozolomide. Consideration of DNA repair pathways, p53-mediated apoptosis and autophagy, convergent proliferation pathways, and epigenetic mechanisms demonstrate avenues for the development of sophisticated drug targets and combination treatments. Ultimately, this article highlights each of these mechanisms and presents referential material for future endeavors in organoid-based drug screening.
Introduction
Glioblastoma (GBM) is one of the most malignant cancers and aggressive tumors, with a rapidly increasing global incidence rate, devastating prognosis, and a low median survival rate of only 9 to 12 months [1]. Classified as a grade IV astrocytoma, GBM is the most common brain cancer, making up 60% of adult brain tumors. Current standard treatment relies on a rigorous combination of total surgical tumor resection, radiation, and concurrent and adjuvant chemotherapy with the DNA alkylating agent temozolomide (TMZ), the first and most widely used chemotherapeutic drug for GBM to date. With an originally promising mechanism, TMZ functions by targeting single strands of DNA and methylating DNA purines at specific positions, resulting in the insertion of incorrect base pairs. This ultimately creates irreparable mutations that lead to single-stranded and double-stranded DNA breaks and halts the cell cycle at the G2/M checkpoint or leads to apoptosis [2]. Despite these efforts, TMZ has unfortunately remained unsuccessful in over 50% of GBM patients, presenting an unresolved complex challenge for researchers to tackle: GBM’s notorious resistance and recurrence [2]. Over the past decades, researchers have focused on a variety of mechanisms for overcoming GBM’s resistance to all forms of therapy. Factors characterizing GBM’s rapidly evolving resistant nature, such as its cellular hierarchy, high heterogeneity, and recurrent genetic and molecular abnormalities, enable its initiation, propagation, unchecked proliferation, resistance, and (worst of all) metastasis. In the clinic, patients who have GBM suffer most from tumor recurrence. The only options for managing recurrence are re-radiation, multiple rounds of systemically administered chemotherapy, experimental immunotherapies, and multiple rounds of surgery, which all ultimately prove futile. Therefore, it is of vital importance to understand how chemo- and radiation-resistance in GBM and its subpopulations enable the complexity and uncontrolled invasiveness of this disease.
Rare populations such as glioblastoma stem cells (GSCs) and circulating tumor cells (CTCs) share unique epigenetic and genetic gene mutations that contribute to GBM’s chemoresistance, radiation-resistance, tumorigenesis and metastasis [3]. GSCs, in particular, possess stem-cell renewal pathways and epigenetic signatures that proliferate and enrich GBM with a subpopulation of hyper-resistant cells [4]. On a broad level, this includes upregulation of DNA repair machinery, increased chromatin methylation, and proliferative mechanisms. These features, in addition to the cross-talk between numerous other signaling pathways, ultimately contribute to TMZ treatment failure and the inevitable recurrence of GBM.
A series of studies have shown the role of signaling pathways in the development of resistance in GBM. A signaling pathway encompasses a series of chemical reactions in which a group of molecules in a cell work together to control a cellular function. From DNA repair pathways, such as the O6-methylguanine-DNA methyltransferase (MGMT) or base excision repair (BER) pathways, to apoptosis pathways involving p53 and caspases, signaling pathways are a key target to understanding how to overcome the challenges of GBM treatment. Furthermore, the interactions between numerous signaling pathways have been discovered to play a crucial role in TMZ resistance. The cross-talk activation of receptor tyrosine kinases (RTKs), such as epidermal growth factor receptor (EGFR) and mesenchymal-epithelial transition (MET) factor, among other pathways, contribute to oncogenic switching and increased proliferation signal persistence [5]. Moreover, numerous epigenetic mechanisms have been implicated in TMZ resistance, including repressive DNA binding, chromatin remodeling, microRNAs (miRNAs), and long non-coding RNAs (lncRNA). In this review, we explore the different mechanisms of GBM signaling pathways that have the highest potential to combat GBM resistance and present promising avenues of treatment to target drug resistance.
Ethics statement: This study was a literature review of previously published studies and was therefore exempt from institutional review board approval.
Treatment challenges
TMZ, the current therapeutic drug for GBM, methylates DNA as a way of targeting and destroying GBM cells through DNA mismatch accumulation, inducing cell cycle arrest at G2/M and apoptosis. TMZ is a prodrug of the anti-cancer drug TMZ, and an imidazotetrazine derivative of the alkylating agent dacarbazine (Fig. 1). With these properties and its lipophilic nature, TMZ is able to cross the blood-brain barrier, and effectively create nucleotide lesions [6]. Specifically, TMZ induces cytotoxic O6-MeG lesions via methylation at the O6 position of the guanine nucleotide [7]. Alternatively, radiation therapy damages DNA in GBM cells via photon-induced single-stranded and double-stranded DNA breakage inducing a widespread DNA damage response [8]. However, GSCs and other resistant populations can easily overcome these mechanisms by activating their DNA damage checkpoints and other compensatory signaling pathways. Furthermore, the enzyme MGMT is enriched in chemoresistant GBM and is specifically responsible for repairing the cytotoxic TMZ-induced DNA lesions in GBM cells. This ultimately weakens TMZ and radiation therapy, as they become ineffective due to resistant counteractivity from GBM subpopulations that express features of GSCs, MGMT, cross-talking signaling pathways, and other epigenetic mechanisms. Thus, it is important to further investigate these mechanisms at a deeper level to create more effective methods of overcoming the barrier of therapeutic resistance in the management of GBM.
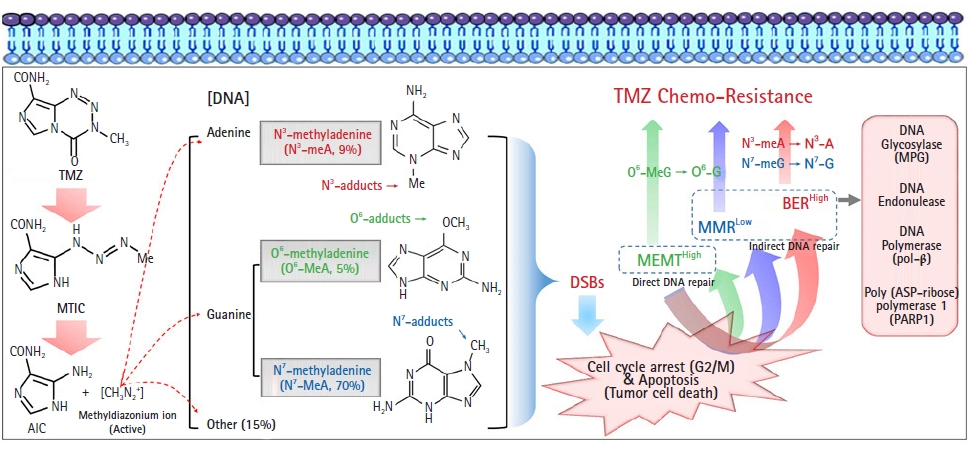
Types of DNA damage by temozolomide (TMZ) and DNA repair mechanisms involved in its resistance in glioblastoma. An active ion (methyldiazonium cation) converted from TMZ transfers a methyl group to DNA, subsequently leading to cytotoxic DNA lesions such as methylation of DNA guanine residues at the O6 (O6-MeG, about 5% of all DNA lesions) and N7 (N7-meG, about 70% of all DNA lesions) positions, as well as adenine at the N3 position (N3-meA, about 9% of all DNA lesions). O6-MeG is initially repaired by active MGMT, and unrepaired O6-MeG is continuously repaired by indirect DNA repair mechanisms, such as the MMR system. The MMR system will mispair with thymine on unrepaired O6-MeG, and genomic instability will be amplified through several cycles of futile MMR. Another indirect DNA repair mechanism is base excision repair (BER). N7-meG and N3-meA adducts can be effectively removed by the BER system through mutual cooperation with components such as methylpurine DNA glycosylase (MPG), DNA endonuclease, DNA polymerase (pol-β), and poly(ADP-ribose) polymerase (PARP). MTIC, methyltriazine imidazole carboxamide; AIC, aminoimidazole carboxamide; DSB, double-strand break; MGMT, methylguanine methyltransferase.
DNA repair pathways
1. MGMT
MGMT is one of the main enzymes that is tied to conventional TMZ resistance. Even though, TMZ functionally leads to the alkylation of the N7 guanine and N3 adenine more than the O6 guanine nucleotide position, the activity and expression profile of MGMT at removing O6-MeG lesions is highly correlated with TMZ resistance status in close to 50% of patients [7]. This is tied to the highly cytotoxic nature of O6-MeG lesions and the alternative repair mechanisms for N-targeted lesions [2]. Since MGMT directly opposes TMZ activity, research in the past decade has focused on the expression mechanics of MGMT in TMZ-resistant GBM. Particularly, the methylation status of the promoter site in the cysteine phosphate-guanine (CpG) MGMT gene locus directly correlates with the MGMT expression level and TMZ resistance within GBM. When this site is methylated, MGMT expression is low and GBM is TMZ-sensitive; when the site is unmethylated, MGMT expression is high and GBM is TMZ-resistant.
However, this relationship is not fully linear, as there exist many TMZ-resistant GBM lines that have methylated MGMT promoters and TMZ-sensitive lines with a hypomethylated MGMT promoter. More importantly, some TMZ-resistant GBM cells do not express MGMT [9–11]. For this reason, researchers have begun to elucidate convergent and parallel signaling pathways that contribute to GBM chemoresistance, including other DNA repair pathways such as BER and mismatch repair (MMR), chromatin remodeling mechanisms, the overall maintenance of a stem-cell state, and tumor microenvironmental factors (Fig. 1) [2]. Ultimately this suggests that TMZ resistance is not simply based on the activity level of MGMT, but that there exist alternative mechanisms. Throughout this review, we continue to identify these alternative mechanisms for TMZ resistance.
2. MMR
TMZ-induced DNA methylation leads to the creation of toxic lesions and mismatches during DNA replication. An O6 Me-G lesion can lead to an O6 methylguanine to thymine mismatch, which can result in activation of the MMR mechanism. At the time of DNA replication, this can lead to a double-strand DNA break via repeated attempts to replace the mismatched thymine nucleotide. Repetition of these DNA breaks induced by the MMR system in response to TMZ-based lesions results in G2/M arrest and eventually apoptosis (Fig. 1) [12]. Based on this mechanism, it is clear that the effectiveness of translating TMZ-induced cytotoxic lesions into cell arrest or apoptosis hinges on an intact MMR system. Mutations in the MSH6 gene of the MMR system have been implicated in TMZ-resistant GBM. TMZ-induced mutations themselves within MMR genes, as a result, could be a potential mechanism for acquired TMZ resistance [13]. Moreover, the expression pattern of MMR proteins seems to be in opposition to the activity of MGMT, where low MGMT activity and high MMR activity are linked to high TMZ sensitivity. Based on this feature, it may be pertinent to reinduce or stimulate MMR activity to enhance the accumulation of cytotoxic DNA breaks, apoptosis and, ultimately, TMZ sensitivity [14].
3. BER
BER is a key alternative pathway to repair single-nucleotide lesions. TMZ specifically creates the majority (90%) of its lesions at the N-sites of nucleotides such as the N7-G or the N3-A lesion, and BER conventionally and rapidly repairs these lesions, leading to GBM survival. Within the BER mechanism exist numerous enzymes, including DNA glycosylase, endonuclease, polymerase, and DNA ligase, whose activity can impact GBM resistance to TMZ (Fig. 1). Specifically, DNA glycosylase MPG, stem cell factor high-mobility group A2, and DNA polymerase-β (pol-β) have been shown to contribute to TMZ resistance via mechanisms of DNA repair (Fig. 1) [15,16]. Moreover, poly(ADP-ribose) polymerase 1 (PARP1) has been shown to recognize single-stranded DNA breaks and protect DNA fragments from accumulation (Fig. 1). Based on this observation, the elimination of the pol-β of BER in combination with a PARP1 inhibitor has been proposed to be effective in enhancing TMZ sensitivity by increasing the accumulation of damaged DNA [16,17].
Apoptosis, p53, and autophagy
1. p53
p53 is an interesting player in the effectiveness of TMZ. Specifically, p53 is a tumor suppressor gene that is important for the binding and recognition of DNA damage, ultimately stimulating p21, blocking cdk2, and causing cell cycle arrest [18]. Moreover, the activation and accumulation of p53 lead to the direct transcription of BH3-only proteins, which activate apoptosis (Fig. 2) [19]. Since TMZ’s primary function is to create DNA lesions aimed at triggering arrest or apoptosis, mechanisms involving p53 are relevant both to GBM and chemoresistance.
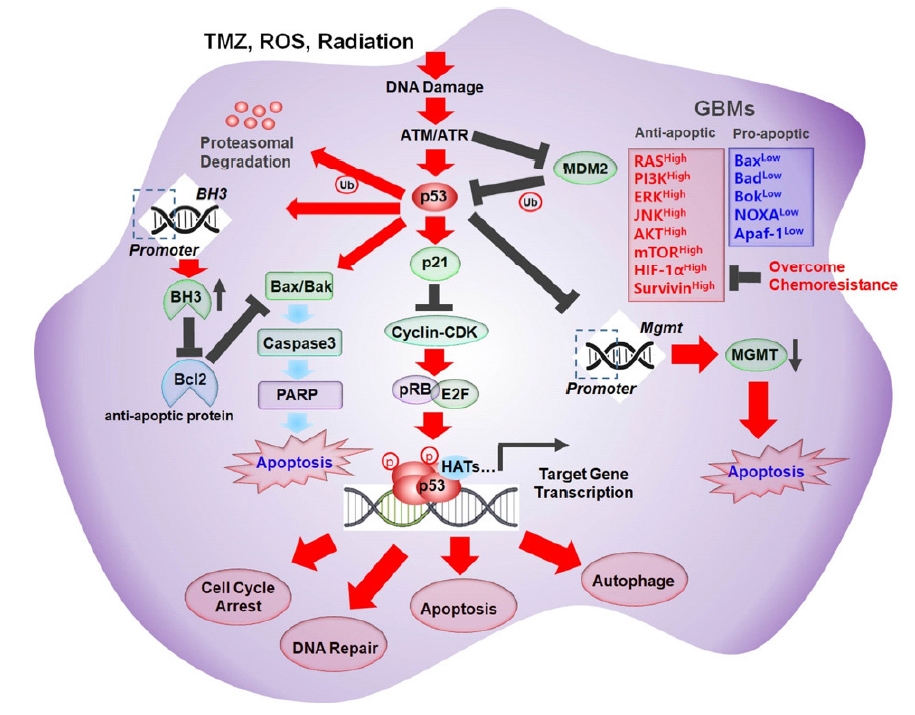
Schematic illustration of the molecular mechanism of various p53-mediated cellular events. Temozolomide (TMZ), reactive oxygen species (ROS), and radiation increased the accumulation of DNA damage which resulted in stimulation of ATM/ATR/p53 signaling pathway. Activated p53 increased expression of p21, and Bax/Bak but down regulated expression of Bcl2, and MGMT, subsequently lead to various cellular dynamics of glioblastoma (GBM) cells. PI3K, phosphatidylinositol 3-kinase; JNK, c-Jun N-terminal kinase; HIF, hypoxia inducible factor; PARP, poly(ADP-ribose) polymerase; MGMT, methylguanine methyltransferase.
Ultimately, most GBM tumors (87%) exhibit some alteration to the TP53 gene or members in its signaling pathways, such as an amplification of the negative regulator MDM2. Upon further investigation, there is no clear consensus on the impact of p53 on TMZ resistance. Some studies have reported that wild-type p53 conferred more TMZ resistance than non-functional p53, but others have reported that TMZ-sensitive GBM cells (e.g., A172, U87) express wild-type p53, and p53 gene mutations were found in TMZ-resistant GBM cells (e.g., LN-18, T98G, U138) as well as TMZ-sensitive GBM cells (e.g., U251, U373) [7].
Beyond these points, there is a clear connection between p53 and the expression of MGMT. Numerous researchers have confirmed that p53 is a negative regulator of MGMT gene expression and can create an MGMT-depleted state (Fig. 2). Based on this observation, reactivation of wild-type p53 is a potential mechanism to overcome TMZ resistance in MGMT-overexpressing GBM. MDM2, as mentioned before, negatively regulates p53 by targeting p53 for proteasomal degradation (Fig. 2). Based on this, a mechanism for re-activating the p53 molecule within GBM involves nutlin-3, which competitively inhibits the binding of MDM2 to p53, allowing for potent p53 activation, MGMT downregulation, and TMZ desensitization [20].
2. Apoptosis
Beyond the activity of p53, the apoptotic signaling pathway is integral to the effectiveness of TMZ and other chemotherapies. Apoptosis is characterized by the activation of internal signaling pathways, due to the accumulation of reactive oxygen species (ROS), DNA damage, and overall cell damage, leading to a convergence on caspase enzymes that degrade the cell into apoptotic fragments (Fig. 2). For TMZ and other GBM therapies, the activation of apoptosis is critical for decreasing tumor bulk. Unfortunately, overall resistance to TMZ and other treatments can be tied to dysregulated apoptotic and anti-apoptotic proteins within GBM [21]. Specifically, the anti-apoptotic proteins Bcl2, Bclx, and Mcl-1, in addition to cell survival proteins including RAS, PI3K, ERK, c-Jun N-terminal kinase (JNK), AKT, mTOR, and hypoxia inducible factor (HIF-1α), have been shown to be overexpressed in GBM (Fig. 2). Moreover, decreased expression of pro-apoptotic proteins, including Bax, Bad, Bok, NOXA, and apoptotic protease activating factor-1 (Apaf-1), has been observed in GBM (Fig. 2) [22]. In addition, TMZ has been shown to bias anti-apoptotic mechanisms by promoting the degradation of Mcl-1, thereby reinforcing Bcl-2/Bcl-Xl-induced resistance to apoptosis [23]. The increase in Bcl2/Bak is reinforced by the PI3K/Akt pathway, which inhibits the pro-apoptotic Bad [24]. Based on these findings, inhibition of Bcl-2 member proteins is a potential method of overcoming TMZ-induced apoptosis resistance. Survivin, another anti-apoptotic protein, has been shown to block TMZ-induced apoptosis, conferring TMZ resistance. Therefore, specific inhibition of survivin is another method of increasing TMZ sensitivity (Fig. 2) [25]. Ultimately, shifting the balance of apoptotic factors is another method of increasing the effectiveness of TMZ in GBM.
3. Autophagy
Autophagy is another cellular process that is relevant to TMZ resistance. Autophagy is the process by which damaged proteins, organelles, and other cellular debris are segregated into autophagosomes and trafficked for lysosomal degradation. Autophagy, thus, can be seen as a process of cellular renewal and a method to bypass TMZ-induced cellular damage and apoptosis [26]. In most cases, this feature of autophagy is considered a mechanism of TMZ resistance; however, late-stage activation of the autophagy pathway can trigger mitochondrial permeability and reinforce apoptosis and thereby TMZ-mediated cytotoxicity. Therefore, the role of autophagy has been controversial, as it may lead to cancer death or cancer survival. For these reasons, it is important to understand the convergent and cross-talking pathways in autophagy in relation to TMZ resistance or sensitivity [27,28].
Endoplasmic reticulum stress and damage associated with TMZ have been linked to activation of the unfolded protein response, which in turn triggers IRE1 to activate XBP1, ASK1, and JNK, promoting autophagy and apoptosis (Fig. 3) [29]. Similarly, TMZ has been shown to activate ATM/AMPK/ULK1-mediated autophagy [30]. In TMZ-resistant lines, AMPK inhibitors have been shown to downregulate TMZ-induced GBM cell apoptosis [31]. For this reason, ATM/AMPK/ULK1 is another pathway relevant to autophagy-targeted methods of overcoming TMZ resistance (Fig. 3) [32]. In addition, activation of the PI3K/Akt/mTOR pathway via RTK activity and TMZ-induced DNA damage is linked with autophagy (Fig. 3). mTOR, in particular, is noted as a key regulator of autophagy, so selective mTOR inhibition has also been tested as a method to downregulate TMZ resistance-associated autophagy [33]. The CaMKKβ/AMPKα/mTOR pathway has been implicated in upregulating TRPC5, an initiator of autophagy, leading to TMZ resistance. For this reason, selective inhibition of mTOR-associated TRPC5 is another target to overcome TMZ resistance [34].
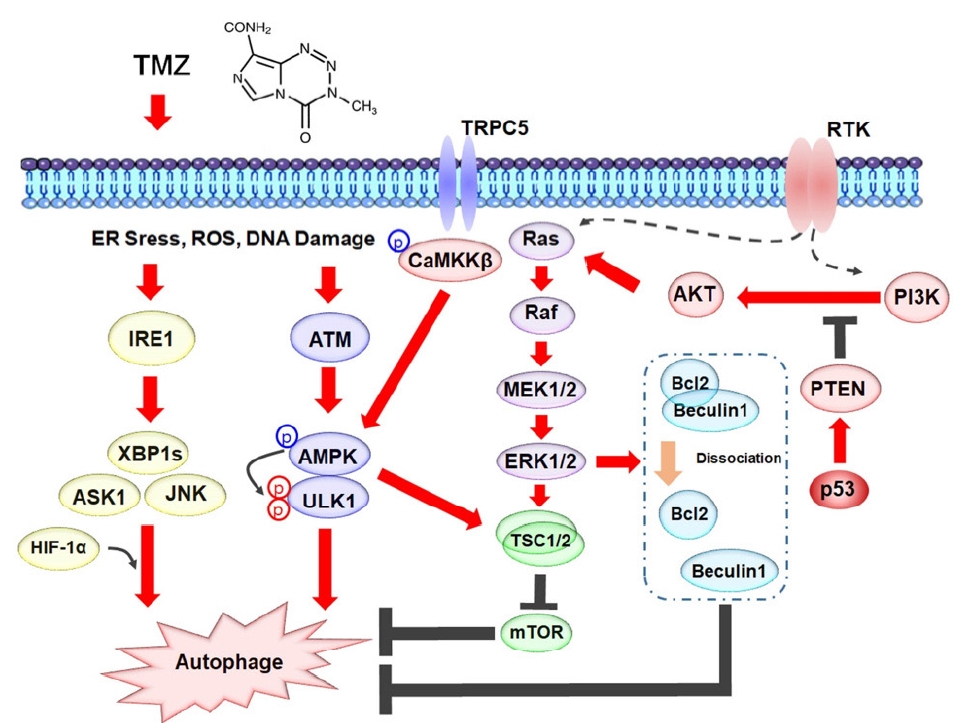
Signaling pathways stimulated by temozolomide (TMZ) are involved in autophagy in glioblastoma cells. A schematic model shows TMZ induces endoplasmic reticulum (ER) stress, reactive oxygen species (ROS), and DNA damage result in autophagy through unfolded protein response signaling, TRPC5/CaMKKβ/AMPK signaling, Ras/MAPK/mTOR signaling, and PI3K/AKT/MAPK signaling pathways. RTKs, receptor tyrosine kinases; PI3K, phosphatidylinositol 3-kinase; IRE1, inositol-requiring enzyme 1; XBP1, X box-binding protein 1; JNK, c-Jun N-terminal kinase; HIF, hypoxia inducible factor.
The accumulation of ROS and TMZ-induced DNA damage, as well as pathogenic RTK activation, is also known to activate the MAPK pathway and initiate autophagy [35]. The relevance of this particular MAPK/ERK/MEK axis to TMZ resistance is somewhat unclear. It has been found that ERK1/2 is hyperactivated in TMZ-resistant lines, thereby inhibiting apoptosis tethered to late-stage autophagy via the ERK1/2/Bcl-2/Beclin-1 axis. Moreover, in these TMZ-resistant lines, some studies have observed decreased autophagy. Specifically, MAPK and ERK1/2 can promote the activation of Bcl-2 transcription, thereby inhibiting the induction of autophagy via the dissociation of the Bcl-2/Beclin-1 complex (Fig. 3). Along these lines, late-stage induction of autophagy via the berberine, which reduces ERK1/2 signaling, has been shown to overcome TMZ resistance [36]. These observations are still a bit counterintuitive, but strongly suggest the importance of MAPK signaling in regulating autophagy in TMZ-resistant GBM.
Furthermore, numerous other signaling pathways have been tied to TMZ-induced autophagy and TMZ resistance including Wnt/β-catenin and JAK/STAT signaling. With mixed results, autophagy-targeted methods nonetheless show substantial promise for overcoming TMZ resistance.
Cross-talk in signaling pathways
1. RTKs
RTKs are well-conserved signaling transduction pathways that are often dysfunctional and malignantly amplified in cancer. In particular, the insulin-like growth factor receptor (IGFR), hepatocyte growth factor receptor (HGFR; also known as MET), fibroblast growth factor receptor (FGFR), vascular endothelial growth factor receptor (VEGFR), and most importantly the EGFR family, have been shown to be highly correlated with GBM chemoresistance. Moreover, the downstream pathways of RTKs, Ras/MAPK/ERK and Ras/PI3K/AKT, which have mutations in close to 90% of GBM tumors, both have cross-talk and impacts on cell survival, TMZ-resistance mechanisms, and overall proliferation [37]. Focusing on AKT, several downstream targets are implicated in TMZ resistance. Pyruvate dehydrogenase kinase 1 (PDK1), HIF-1, forkhead box O3 (FoxO3a), and NF-κB are all key factors in Akt-mediated chemoresistance (Fig. 4). HIF-1 and its related proteins HIF-1α and HIF-1β are both activated by Akt and, in turn, activate vascular endothelial growth factor (VEGF), which is critical for tumor angiogenesis (Fig. 4). Modulation of VEGF expression is another method of targeting TMZ-resistant GBM (Fig. 4) [2]. The continuous expression of NF-κB in resistant GBM is linked to the pro-survival mechanism, co-activation of ATM pathways, and even upregulation of MGMT [38]. Overall, these features make NF-κB another target for overcoming TMZ resistance.
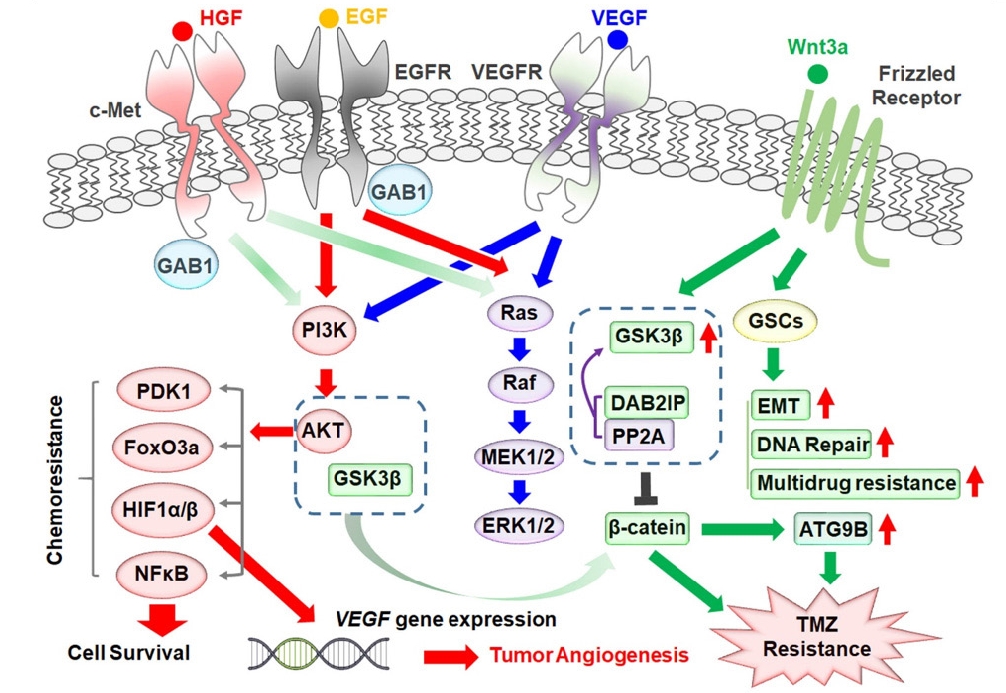
Cross-talk between receptor tyrosine kinase signaling pathways and the Wnt/β-catenin signaling pathway contributing to temozolomide (TMZ) chemoresistance in glioblastoma cells. HGF, hepatocyte growth factor; EGF, epidermal growth factor; EGFR, EGF receptor; VEGF, vascular endothelial growth factor; VEGFR, VEGF receptor; GAB1, growth factor receptor-bound protein 2 associated binding protein 1; PI3K, phosphatidylinositol 3-kinase; PDK1, pyruvate dehydrogenase kinase 1; HIF, hypoxia inducible factor; DAB2IP, DAB2 interacting protein; GSCs, glioblastoma stem cells; EMT, epithelial–mesenchymal transition.
EGFR in particular is a prominent receptor and pathway that is mutated and often overexpressed in GBM, leading to ligand-independent activation of both the Ras/MAPK/ERK and Ras/PI3K/AKT pathways. Moreover, the EGFRvIII mutation, which is found in close to 50% of cells, malignantly activates MAPK and AKT but also has been shown to increase TMZ sensitivity by upregulating MMR in MGMT-silent tumors [39]. Based on these observations, simple EGFR inhibition may lead to counterintuitive effects regarding TMZ resistance. Moreover, cross-talk between EGFR and MET has been recently identified as a method of oncogenic switching and chemoresistance. In particular, both EGFR and MET share the downstream activation of P13K and recruitment of the GAB1 adapter (Fig. 4). With many resistant GBMs possessing EGFR mutants that are constitutively active, GAB1 can preferentially associate with EGFR. Since both MET and EGFR are capable of cross-talk, selective inhibition of EGFR is ineffective at downregulating the PI3K pathway due to cross-talk recruitment of GAB1 by MET activation (Fig. 4). This feature, also known as oncogenic switching, is a common feature of RTKs and contributes to malignant signal persistence and overall chemoresistance. Based on this observation, some studies have reported success in the use of dual-functionalized EGFR/MET nano-inhibitors [5].
Based on these overall observations, methods of targeting RTKs to overcome chemoresistance, must take into consideration downstream cross-talk within and between both MAPK/ERK and AKT/PI3K signaling pathways.
2. Wnt/β-catenin
Wnt/β-catenin pathways are central signaling pathways that have wide-scale impacts on the behavior of chemoresistant GBMs. With both a canonical (β-catenin dependent) and a non-canonical (β-catenin independent) pathway, Wnt signaling is integral to the development of GSCs, the maintenance of stemness, the induction of upregulated DNA repair pathways, the induction of cytoprotective autophagy, and overall TMZ resistance (Fig. 4). Moreover, the downstream components of Wnt signaling such as GSK3β and CK1α engage in cross-talk with pathways such as Akt and even epigenetic mechanisms involving repressive chromatin maintenance.
Focusing on the development of GSCs, upregulated Wnt signaling is associated with the induction of epithelial to mesenchymal transitions (EMTs), upregulated DNA repair, and expression of chemoresistance mediators, such as multidrug resistance associated protein-1 (MRP1/ABCC1) (Fig. 4). Wnt3a, in particular, has been found to induce stemness and serves as an effective inducer of TMZ resistance in GBM [40]. Regarding DNA repair, Wnt inhibitors, such as salinomycin, celecoxib, and Wnt-C59, have been shown to decrease the expression of MGMT in GSCs and reintroduce TMZ sensitivity [41]. In terms of relevance to autophagy, Wnt/β-catenin activation has been shown to upregulate ATG9B expression, which is associated with TMZ-resistant GBM autophagy. Inhibition of canonical Wnt inhibits ATG9B expression and overcomes TMZ resistance. This pathway, in particular, involves cross-talk at the GSK3β level, where DAB2 interacting protein (DAB2IP) recruits PP2A and activates GSK3β to inhibit Wnt/β-catenin (Fig. 4). In TMZ-resistant lines, it was found that loss of DAB2IP led to Wnt-mediated activation of protective autophagy [42].
Turning to other cross-talk mechanisms, we can also focus on the interactions of GSK3β with AKT signaling pathways. Stearoyl-coenzyme A desaturase 1 (SCD1), another protein upregulated in TMZ-resistant GBM cells, was found to increase cell proliferation and TMZ resistance through an AKT/GSK3β/β-catenin axis (Fig. 4). Specifically, inhibition of SCD1 was found to promote GSK3β activation and β-catenin degradation, which enabled GBM cells to overcome TMZ resistance [42]. Ultimately, targeting and inhibition of Wnt and downstream targets are a very promising direction for effective combination therapies for TMZ-resistant GBM.
Epigenetic mechanisms
1. MBD3 and GSC maintenance
GSCs are thought to arise from isolated and mutated populations of neural progenitor cells (NPCs). GSCs are theorized to share numerous parallel developmental pathways with NPCs including differentiation, and this idea has been considered for targeted therapies. Beyond this, it is well established that stem cells and cancer stem cells possess a heavily repressive chromatin environment. In particular, methyl CpG binding domain protein 3 (MBD3), which is a core component of the repressive nucleosome remodeling and deacetylase (NuRD) complex, is a key factor in the maintenance and self-renewal of GSCs (Fig. 5). The numerous self-renewal components of GSCs are strongly correlated with high MGMT expression and DNA-repair mechanisms that contribute greatly to TMZ resistance. Based on this concept, we have previously attempted to disrupt MBD3 via proteasome-mediated degradation by activating CK1α kinase. By removing MBD3 from GBM cells and GSCs with high MGMT expression, we have observed the induction of GSC destabilization and differentiation, leading to a loss of GSCs and overcoming TMZ resistance (Fig. 5) [4]. Interestingly, utilizing pyrvinium pamoate (PyrPam), a CK1α activator, can lead to cross-talk and inhibition of the Wnt/β-catenin pathway mentioned above. Canonical Wnt signaling has been heavily implicated in the self-renewal and EMT pathways within GBM and its inhibition via CK1α-mediated phosphorylation, and ubiquitination of β-catenin can lead to an effective target for GBM [43]. Based on these findings, effective activators of both GSC differentiation, and disruption of GSC self-renewal using small molecules like PyrPam may be an effective method to target both epigenetic and conventional signaling pathways and overcome chemoresistance.
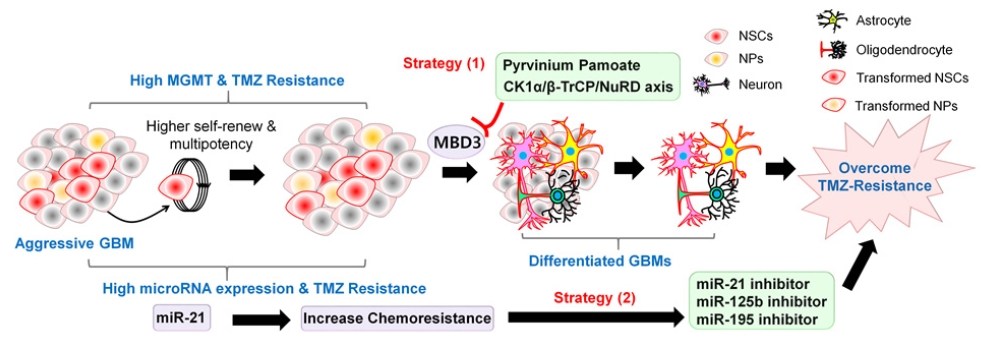
Two different therapeutic strategies to overcome temozolomide (TMZ) chemoresistance in glioblastoma (GBM). Strategy 1: Hypothetical graphical model showing that activation of CK1α-β-TrCP-Mbd3-NuRD signaling induces neural differentiation of glioblastoma stem cells (GSCs) and lead to decrease GBM recurrence and growth. Strategy 2: Downregulation of miR-21, miR-125b, and miR-195 enhances the pro-apoptotic efficacy of TMZ and finally overcome TMZ chemoresistance in GBM treatment. MGMT, methylguanine methyltransferase; NSCs, neural stem cells; NPs, neural progenitors.
2. Histone deacetylases and similar targets
As mentioned above, GSCs self-renew by maintaining a repressive chromatin state. Histone deacetylases (HDACs) cleave acetyl groups of chromatin, leading to the compaction and repression of chromatin regions. Moreover, expression of mutated HDACs has been correlated with the progression of GBM and TMZ resistance. At a conventional level, open chromatin environments are favorable to the alkylation and DNA-damage mechanisms of TMZ, so the HDAC-mediated loss of an open chromatin state directly relates to TMZ effectiveness. More specifically, HDAC8 inhibition has been shown to downregulate MGMT via the ADRM1 proteasome mechanism [44]. Similarly, HDAC6 inhibition has been found to increase MMR proteins while blocking abnormal EGFR and p53 activity [45]. Finally, HDAC1 and HDAC3 inhibition has been shown to aid in overcoming TMZ resistance through the inhibition of NF-κB pro-survival genes [46]. Overall, HDAC inhibition leads to a reinforcement of DNA damage pathways, while inhibiting the pro-survival and DNA repair features of TMZ-resistant GBM.
3. miRNAs
In the past decade of research, the importance of non-coding miRNAs has been investigated in the context of GBM and chemoresistance. Importantly, miRNAs can lead to the degradation and post-translational repression of proteins relevant to tumor progression and cell death. In GBM, it has been found that miR-21 promotes chemoresistance via a decrease in the Bax/Bcl2 ratio and caspase-3 activity, hindering overall TMZ-induced apoptosis (Fig. 5) [47]. Moreover, numerous other miRNAs have been linked to the inhibited suppression of MGMT, leading to the characteristic high MGMT expression in GSCs. Since numerous miRNA expression profiles have been implicated with the signaling pathways mentioned above, including STAT, MGMT, and caspases, the aberrant miRNA profiles of GBMs present pertinent targets to overcome TMZ resistance. Researchers have substantiated these claims through the discovery of miR-195, miR-125b, miR-21 inhibitors that improve the effectiveness of TMZ (Fig. 5) [48,49]. Moreover, supplementation with miR-130-p and miR-204 has been shown to increase TMZ effectiveness via degradation of Sp1 and fibroblast activation protein, respectively [50,51].
4. Long non-coding RNA
In addition to miRNAs, there are many other non-coding RNA mechanisms within GBM that can be tied to TMZ chemoresistance. lncRNAs are RNA transcripts longer than 200 nucleotides that originate from regions that were previously considered transcriptional noise. These transcripts can encompass intron, exon, and promoter regions in both a sense and antisense direction and have multiple functionalities as a result. In the past few years, these lncRNAs have been implicated in numerous mechanisms such as chromatin remodeling, transcriptional regulation, mRNA stabilization, and sequestration and sponging of miRNAs.
In relation to chemoresistance, TMZ-resistant GBM cells have been shown to secrete and be enriched in lncRNA SBF2-AS1, which has been demonstrated to sequester miR-151a-3. This ultimately causes an upregulation in X-ray repair cross-complementing protein 4 and increased DNA repair capabilities, in opposition to the effects of TMZ [52]. In addition, the lncRNA TP73-AS1 has been found to be enriched in GBM samples, upregulating the cancer stem cell marker ALDH1A and contributing to stemness (Fig. 5). When researchers silenced TP73-AS1 in GSC lines, they observed increased sensitivity and cell death with TMZ (Fig. 6) [53]. In relation to the MGMT and GSK3β pathways, the lncRNA MALAT1 has been shown to be enriched in GBM, sponging miRNA101 and upregulating MGMT and GSK3β, directly contributing to conventional TMZ resistance (Fig. 6) [54]. More recently, lncRNA ADAMTS9-AS2 has been shown to promote TMZ resistance via disruption of FUS/MDM2 ubiquitination. The lncRNA ADAMTS9-AS2 has been shown to be enriched in TMZ-resistant GBM and to competitively inhibit FUS degradation via MDM2, leading to increased proliferation and metastatic characteristics. Ultimately, knockdown of lncRNA ADAMTS9-AS2/FUS signaling increases TMZ sensitivity in GBM (Fig. 6) [55]. Similarly, the lncRNA SNHG12 has been associated with TMZ resistance. SNHG12 has been shown to serve as a sponge for miR-129-5p, leading to upregulation of MAPK1 and E2F7, conferring TMZ resistance by promoting the G1/S transition and inhibiting cell apoptosis (Fig. 6). Similarly, hypomethylation at the BSP1 site of the SNHG12 promoter and the activity of SP1 have been tied to the overexpression of SNHG12 and, ultimately, TMZ resistance [56]. With the mounting evidence of many lncRNAs relevant to TMZ resistance, selective lncRNA inhibition shows great potential as a new therapeutic target.

Types and mechanisms of long non-coding RNAs (lncRNAs) causing temozolomide (TMZ) chemoresistance in glioblastoma cells. GSCs, glioblastoma stem cells; MGMT, methylguanine methyltransferase.
Organoid and drug discovery
1. Three-dimensional cerebral organoids
Following the development of human induced pluripotent stem cell (hiPSC)- or human embryonic stem cell-derived 3-dimensional (3D) in vitro culture technology, various mini-organs that can recapitulate the structure and function of the corresponding human organs have been successfully generated [57]. The cerebral cortex occupies almost 70% of the whole brain, and the generation of a 3D cerebral mini-brain and its application for drug screening can be considered a major breakthrough in drug development for neurological disease treatment [58]. However, the early cerebral brain organoid model had limitations, including a deficiency of brain immune cells (e.g., microglia) and blood vessels for the structure of the blood-brain barrier [59]. Scientists have reported an improved hiPSC-derived 3D cerebral organoid with similar tissue structures and functional capacities such as genetic and epigenetic signatures, electrical oscillations, and stimulation-responses to those seen in the human cerebrum (Fig. 7) [60]. This advanced 3D cerebral organoid model, therefore, has been highlighted for various applications in translational research, including in the field of oncology [61].

Establishment of human induced pluripotent stem cell (iPSC)- and embryonic stem cell (ESC)-derived advanced 3-dimensional (3D) cerebral organoids. An advanced cerebral organoid consists of neurons, glial cells, and blood vessels for constructing the brain blood barrier to overcome limitations of the early cerebral organoid model.
2. 3D brain-cancer assembloid
Many therapeutic agents show significant effects in in vitro 2-dimensional cell culture studies and in vivo xenograft animal model studies, but eventually fail clinical trials [62]. This suggests that current cancer models lack representation of the complex biology of cancer, including cancer-brain interactions, cancer-drug responses, and drug resistance seen in patients [63]. GBM occurs most often in the cerebral hemispheres of the brain. Therefore, a 3D cerebral brain-cancer assembloid model system obtained from growing GBM spheroids inside a human 3D cerebral organoid could be the basis for potentially replacing in vitro cancer cell cultures and xenograft animal models as a powerful platform for reproducible drug discovery. Moreover, current advances in brain organoid culture methods to generate different brain regions, such as the cerebellum and hypothalamus, have contributed to advances in brain assembloid technologies, and array-based 3D brain-cancer assembloid technology using these methods will be promising drug screening platforms for the identification of novel therapeutics overcoming cancer drug resistance (Fig. 8).

Establishment of an array-based 3-dimensional (3D) brain-cancer assembloid system for high-throughput drug screening. TMZ, temozolomide; GBM, glioblastoma; GSCs, glioblastoma stem cells.
Conclusion
With the numerous features of chemoresistant TMZ populations, including GSCs, and overall tumor heterogeneity, understanding cross-talk in signaling pathways, DNA repair mechanisms, and epigenetic regulation can provide a more multifaceted strategy for researchers to develop effective GBM treatments. The failure of TMZ in close to 50% of patients and the variability of common resistance markers like MGMT also reinforce the necessity for more combination therapies. For patients with recurrent GBM, the development of effective combination therapies that address DNA repair, apoptosis/autophagy, RTK and Wnt signaling, and miRNA and lncRNA will be integral. Moreover, with our greater understanding of GSCs, CTCs, and the tumor microenvironment, methods of halting metastasis, the negative selection of resistance, and the application of 3D brain-GBM cancer assembloid systems as a drug discovery platform will be within reach. With the high severity, mortality, and recurrence rates of GBM, an understanding of all these features will translate to significant improvements in GBM patient outcomes.
Notes
Conflict of interest
Byoung-San Moon has been an editor of Organoid since 2021. No other potential conflict of interest relevant to this article was reported.
Funding
BSM. is supported by the Basic Science Research Program through the National Research Foundation (NRF) funded by the Ministry of Education (2021R1I1A3047154) and by the Ministry of Oceans and Fisheries, Korea (Grant No. 2021-2615). ISK. is supported by the National Research Foundation of Korea, which is funded by the Korean Government (NRF-2018-R1A6A1A-03024314).
Author contributions
Conceptualization: BSM; Formal analysis: AP, EJL, ISK; Funding acquisition: ISK, BSM; Investigation: AP; Methodology: AP, EJL; Project administration: BSM; Resources: ISK; Supervision: BSM; Validation: EJL, ISK; Visualization: BSM; Writing-original draft: AP, BSM; Writing-review & editing: ISK, BSM.
Data availability
Please contact the corresponding author for data availability.