Induced pluripotent stem cell-derived hematopoietic stem and progenitor cells: potential, challenges, and future perspectives
Article information

Abstract
Hematopoietic stem and progenitor cells (HSPCs) are responsible for the lifetime dynamics of hematopoiesis, as they are well known for their self-renewing ability and multipotency to differentiate into all types of blood cells, including both myeloid and lymphoid lineages. However, due to their limited amount and accessibility, there is a strong need to search out alternative methods to produce HSPCs. In this review, we suggest induced pluripotent stem cells (iPSCs) as a new viable source for HSPC production because these cells have the potential to self-renew while being relatively easy to modify. Recent studies have revealed that the recapitulation of definitive hematopoiesis is the key to the successful in vitro production of HSPCs with multilineage potential. Therefore, we summarized recent progress in establishing the generation of definitive HSPCs with high maturity and functionality in vitro. Definitive HSPCs can be used in disease modeling and gene therapy for genetic blood disorders via gene modification in iPSCs, applied in cellular immunotherapy in the form of a universal chimeric antigen receptor system, and may recapitulate the intricate immune system within the iPSC-derived organoids that closely mimic the in vivo pathophysiological environment. In summary, this review provides an overview of the generation of HSPCs from iPSCs, in terms of the developmental process of hematopoiesis, in vitro attempts to produce iPSC-derived definitive HSPCs, and the following applications of these cells in numerous areas. This review sheds light on the concept of iPSC-derived definitive HSPCs, setting a milestone for artificial blood production in the near future.
Introduction
Hematologic diseases are common disorders that primarily affect the blood and blood-forming organs [1]. One possible way to classify them is to sort them according to their cause—red blood cells (e.g., sickle cell disease [SCD] or thalassemia) and white blood cells (e.g., lymphoma, leukemia, or myelodysplastic syndrome), which are the solid parts of our blood system [2]. These disorders are prevalent in our society. For instance, according to the World Health Organization, approximately 5% of the world’s population carries trait genes for red blood cell-related disorders, mainly SCD and thalassemia, while over 300,000 babies are born each year with these disorders [3]. Thus, according to the American Cancer Society, an estimated 60,650 and 89,010 new cases will be diagnosed, while 24,000 and 21,170 people will die from leukemia and lymphoma, respectively, in the United States in 2022 [4].
Hematopoietic stem cell transplantation (HSCT) has long been one of the primary therapeutic options for these hematologic diseases. Here, hematopoietic stem and progenitor cells (HSPCs) are adult stem cells (ASCs) residing within the bone marrow with self-renewing abilities and multipotency. After transfusion into a conditioned recipient, HSPCs may demonstrate long-term engraftment as well as reconstitution of the entire hematopoietic system. The blood components derived from HSPCs include red blood cells that are capable of efficient oxygen transportation, megakaryocytes that may further differentiate into platelets and regulate blood clotting, and immune cells that are responsible for both innate and acquired immunity against antigens [5]. Indeed, the European Society for Blood and Marrow Transplantation reported a continuous rise in both allogeneic HSCT and autologous HSCT over 30 years, reaching 48,512 HSCT cases in 2019 [6]. Allogeneic HSCT is conducted using HSCs from immunocompatible donors, whose cells are healthy and may be prepared beforehand, allowing fast treatment. Meanwhile, autologous HSCTs are performed using the patient’s own HSCs, which lower the risk of hazardous complications including graft-versus-host disease (GvHD), graft failure, and infections. However, allogeneic HSCT is hampered by the paucity of human leukocyte antigen (HLA)-matching donors while autologous HSCT is limited by the scarcity of the conventional sources of human HSPCs in terms of number and accessibility, as transplantable HSCs only account for 1 in 106 cells in the human bone marrow [7]. Thus, the ex vivo expansion of harvested HSPCs is difficult as these cells exhibit limited engraftment capacities and functionalities without bone marrow niches [8]. Therefore, there exists a strong need to find an alternative source to produce a sustainable amount of immunocompatible HSPCs for regenerative medicine.
In recent research, induced pluripotent stem cells (iPSCs) have been offered as a potential source to generate HSPCs. iPSCs are artificially generated embryonic stem cell-like cells that are self-renewing and may differentiate into all 3 germ layers (ectoderm, mesoderm, and endoderm) [9]. They were first demonstrated by Yamanaka Shinya in 2006, who introduced a set of reprogramming factors known as OSKM (OCT4, SOX2, KLF4, and c-MYC) into mouse somatic fibroblasts, then into human somatic cells a year after, using retroviral vectors [10,11]. These iPSCs can differentiate into all types of cells, including HSPCs and further differentiated blood cells. Therefore, they have been utilized for disease modeling by recapitulating the developmental process of hematopoiesis. In addition, cells differentiated into different lineages from iPSCs have enabled more advanced modeling of physiologically relevant organ microenvironments [12], such as the vascular system [13–15], immune system [16,17], and even connecting multiple organs [18,19]. Another important point is that iPSCs can be produced from particular patients while retaining their entire genomes. As they are advantageous to genetic correction of disease-causing mutations, HSPCs derived from patient-specific iPSCs have sered as a promising source for autologous HSCT therapy for treating genetic blood disorders [20,21]. Moreover, “off-the-shelf” immunocompatible iPSCs generated by silencing the genes associated with the immune response after transplantation may also extend the feasibility of allogenic HSCT by allowing ready-to-use products without severe rejection to be prepared [22,23]. Hence, many strategies have been devised to generate HSPCs from this novel source. However, achieving high-yield production of iPSC-derived HSPCs with sustained engraftment potential remains challenging. Thus, robust and reproducible protocols for differentiating iPSCs into HSPCs with high maturity and functionality have been actively developed. In this review, we present an overview of the generation of HSPCs from iPSCs and the applications of these iPSC-derived HSPCs.
Ethics statement: This study was a literature review of previously published studies and was therefore exempt from institutional review board approval.
Generation of HSPCs
1. Developmental process of hematopoiesis
As iPSCs correspond to cells in the inner cell mass of blastocyst-stage embryos, the differentiation of these cells into HSPCs mimics the intrinsic system of blood cell generation (hematopoiesis) during embryogenesis. Therefore, it is worthwhile to understand the full process of in vivo hematopoiesis. Insightful studies have investigated the emergence of HSPCs in multiple species, including zebrafish, and hematopoiesis was mainly deciphered using mouse models, which represent vertebrate hematopoiesis well and have a close resemblance to humans in relevant aspects [24,25].
Blood development is known to occur mainly through 3 successive hematopoietic waves, which are separate in space and time [24–26]. The first wave, also known as “primitive hematopoiesis,” takes place in the yolk sac blood islands at mouse embryonic day 7.5 (E7.5). This wave is known to give rise to various immature blood cells, which collectively play the role of first innate immunity in embryos. It includes the production of large nucleated erythrocytes containing embryonic globin proteins, large and multinucleated megakaryocyte progenitors that produce discoid anucleate platelets, and monocyte-independent macrophages without peroxidase activity that are nonetheless responsible for tissue remodeling and lymphatic development. The transient “pro-definitive” second wave follows, giving rise to erythro-myeloid progenitors at E8.25 and lympho-myeloid progenitors at E8.5. The subsequent onset of cardiac contractions allows the circulation of these cells, which migrate to the fetal liver and are detected as neonatal HSPCs. The third wave, “definitive hematopoiesis,” first takes place in the dorsal aorta of aorta-gonad-mesonephros region and vitellin/umbilical arteries at E10.5, and thereafter in the placenta and extraembryonic tissues. This particular wave differs from the previous 2 waves as it may give rise to the first true HSPCs, known as definitive HSPCs. These definitive HPSCs have robust long-term self-renewing abilities, are capable of colonizing hematopoietic organs, and may differentiate into all hematopoietic lineages with mature morphology and functionality for the entire lifespan. They are mainly distinguished by their maturation into enucleated erythrocytes with fetal-to-adult globin switching ability and all lymphoid lineages including B cells, T cells, and natural killer (NK) cells. HSPCs produced in this stage localize in the fetal liver, colonize the fetal thymus and spleen afterwards, and finally seed in the bone marrow before birth, where they engage in life-long hematopoiesis.
As shown, there exists a great difference in the types of blood cells that are generated between primitive and definitive hematopoiesis. While primitive hematopoiesis may only induce a small amount of immature blood cells with limited functionality, definitive hematopoiesis can induce HSPCs with multilineage differentiation potential, particularly mature erythrocytes and lymphoid cells. Therefore, the future of HSPC production with high therapeutic potential lies in the successful recapitulation of definitive hematopoiesis.
Definitive hematopoiesis for the successful generation of definitive HSPCs is marked by 3 crucial developmental steps: (1) induction of the mesoderm lineage, (2) formation of the hemogenic endothelium (HE), and (3) the endothelial-to-hematopoietic transition (EHT) [20]. Blood generation starts at the gastrulation stage, which takes place around 3 days from E6.25 to E9.5. During gastrulation, single-dimensional epiblast cells organize into a multilayered and multidimensional structure called the gastrula, which consists of the endoderm, mesoderm, and ectoderm [27]. Here, cells entering the hematopoietic lineage are first specified into mesodermal cells through the primitive streak, under the influence of various signals including BMP, activin/nodal and Wnt signaling pathways [28]. Early mesoderm development is marked by a transient marker Brachyury, later followed by kinase insert domain-containing receptor (KDR, also known as VEGFR-2, CD309), a marker crucial for vasculogenesis [29]. This process is followed by the divergence of various types of endothelial cells, among which exist the HE, a type of vascular endothelial (VE) cells with the potential to generate hematopoietic lineage cells [30–33]. Therefore, these cells are also identified by VE-cadherin (CD144) and the platelet endothelial cell adhesion molecule PECAM-1 (CD31), which are well-known endothelial markers [34,35], along with CD34, which defines the progenitors of the hematopoietic lineage [36]. In the EHT, as the final step of definitive hematopoiesis, a gradual cell fate transition takes place for 3 to 4 days by an upregulation of the hematopoietic transcription program including Runx1, Gata2, Scl, Sox17 in a selected group of cells in the HE. This results in a vast morphological change, where these cells break out from the tight junction of neighboring endothelial cells and bud off as round floating clusters of HSPCs [37]. The generated HSPCs not only express CD34 but also leukosilan (CD43), a pan-hematopoietic marker that segregates HSPCs from endothelial cells [38], and CD45, which may be used to mark the maturation of generated HSPCs [39].
2. In vitro hematopoiesis
Based on the above-mentioned hallmarks, intensive efforts have been made to closely mimic the developmental process of definitive hematopoiesis to generate HSPCs from iPSCs in vitro. Taken together, HSPC differentiation from iPSCs is based on the utilization of various growth factors and cytokines at different stages of differentiation, including the BMP4, VEGF, bFGF, and HSC cytokines [40,41]. It is mainly conducted in the form of two-dimensional (2D) monolayer induction [42–47] and 3-dimensional (3D) embryoid body (EB) formation [48–51]. Such 2D monolayer induction may be further divided into the utilization of feeder cells such as OP9 stromal cells [52], and the application of extracellular matrix coatings. Meanwhile, the 3D induction method may be further developed into organoid systems, which are unique self-organizing 3D culture systems that closely recapitulate the features of actual human organs [53].
Table 1 summarizes recent progress in establishing the generation of definitive HSPCs with high maturity and functionality in vitro [16,54–68]. As shown, various small molecules related to hematopoietic development have been employed for the efficient induction of definitive HSPCs under 2D conditions. For instance, TGF-β inhibitor SB431542 and GSK inhibitor CHIR99021 are regularly used supplements for the successful induction of definitive HSPCs. Ruiz et al. [54] stated that these 2 molecules promote the development of arterial vascular endothelium and the subsequent production of 56.0% of CD43+CD45+ HSPCs, which may produce mature erythrocytes with adult globin. Using the same molecules, Netsrithong et al. [55] also generated 54.1% of CD34+CD43+ floating HSPCs that may differentiate into erythroid and T cell lineages. Meanwhile, Chang et al. [56] produced 41.6% of CD43+CD45+ HSPCs through CD34+SOX17+ endothelium by manipulating canonical Wnt signaling through the treatment of CHIR99021. Furthermore, the aryl hydrocarbon receptor (AhR) was also found to play a fundamental role in hematopoiesis. Leung et al. [57] argued that the suppression of AhR also promoted the expansion of differentiated HSPCs via p21 inhibition. By treatment with the AhR antagonists CH-223191 and CHIR99021, they induced two-fold expansion of CD34+CD45+ HSPCs that showed high myeloid potential and thus may form adult-type erythroblasts. Therefore, this strategy might contribute to the massive production of iPSC-derived HSPCs with high maturity and functionalities. Moreover, Uenishi et al. [58] demonstrated the role of Notch signaling in the specification of definitive CD144+CD43-CD73-DLL4+ HE by treatment with the Notch activator DLL1-Fc. HSPCs produced from these HE cells showed differentiation abilities toward T cells and erythrocytes expressing enhanced adult-type globin.
In vitro differentiation of human pluripotent stem cells into definitive HSPCs
Aside from the above-mentioned 2D induction methods, various forms of 3D models are also widely used alongside these small molecules to induce definitive HSPCs with multilineage potential. For instance, Sturgeon et al. [59] succeeded in specifying human iPSC-derived EBs into KDR+CD235a- definitive progenitors and, subsequently, 55.0% of CD144-CD45+ HSPCs by using the activin-nodal signaling antagonist SB431542 and the Wnt agonist CHIR99021 at the early stage of development. The cells produced through this method were capable of differentiating into erythrocytes expressing adult globin and lymphoid lineages including NK cells and T cells. Moreover, Angelos et al. [60] demonstrated a significant increase in the total percentage of CD34+CD31+ HE cells (48.2%) along with a two-fold increase in the CD34+CD45+ HSPCs (36.7%) by the treatment of AhR antagonist StemReginin-1 or CRISPR knockout of AhR in a 3D EB model. The cells induced through this method were also able to differentiate into NK cells. There are also variations regarding the types of 3D models. For instance, Flippe et al. [61] induced 37.0% of CD34+CD45+ HSPCs through the utilization of an EB model, while these cells were used for further differentiation of T cells and NK cells through coculture with the Notch ligands OP9-DLL1/DLL4. Meanwhile, Haro-Mora et al. [62] developed a 3D iPS-sac model, a hemangioblast-like structure that contains long-term CD34+CD38-CD90+CD45RA-CD49f+ HSPCs from which mature erythrocytes emerged. Ackermann et al. [16] also established a 3D organoid-like differentiation system (referred to as a humanoid), which showed the defined stages of early hematopoietic development. Subsequently, the EHT through the coculture of hemanoid-derived CD34+CD144+ HE cells with OP9-DLL1/DLL4-expressing stroma cells in IL-3 supplemented medium allowed the successful generation of CD43+CD45+ HSCs (68.3%) with multilineage potential, including macrophage and granulocytes. Thus, Motazedian et al. [63] combined swirler culture where bulk cell-aggregation into EB takes place along with an air-liquid interface culture to produce lymphopoietic organoids expressing extensive network of vascular structures. Here, RAG1-GFP pluripotent stem cells were used for production, where RAG1 is a gene that mediates T cell receptor and B cell immunoglobulin gene rearrangements. RAG1+ HSPCs emerging from the HE of this organoid showed multipotency, and could be further differentiated into myeloid, erythroid, B cells, and T cells. Montel-Hagen et al. [64] recently introduced an enhanced 3D model (the 3D artificial thymic organoid system) that induced definitive CD34+CD43+CD45+/- HSPCs with mature T cell differentiation ability. Last but not least, Tamaoki et al. [65] performed scalable HSPC production alongside the elucidation of hemato-vascular ontogenesis by the formation of yolk sac-like organoids. This hematopoietic spheroid system was formed by co-culturing human bone marrow-derived mesenchymal stem/stroma cells and small EBs (100–150 cells). Its structure highly resembled that of the yolk sac, which allowed the large-scale production of CD34+CD43+ HSPCs with multipotency in the absence of exogenous cytokines or growth factors.
Meanwhile, many attempts have been made to promote the engraftment efficiency of differentiated HSPCs by modulating certain genes that confer to the HSPC’s engraftment, self-renewal, and multilineage capacity. For instance, Sugimura et al. [66] transduced 7 transcription factors (ERG, HOXA5, HOXA9, HOXA10, LCOR, RUNX1, and SPI1) into the HE. After interfemoral injection of HE cells and subsequent 2-week treatment of doxycycline in immune-deficient NOD/LtSz-scidIL2Rγnull (NSG) mice, increased chimerism of human CD45+ cells in the peripheral blood of injected mice was observed up to 12 weeks. Thus, human HSPCs (CD34+), erythroid cells (GLY-A+), myeloid cells (CD33+), B cells (CD19+), and T cells (CD3+) were detected in the bone marrow and thymus at week 12, implying the multilineage-engraftment ability of these cells. In addition, Shim et al. [67] reported the activation of these transcription factors by the treatment of HDAC inhibitor SAHA. This resulted in a two-to five-fold increase in the population of CD34+CD45+ HSPCs (58.1%) by committing iPSCs into definitive CD34+CD90+ HE (66.0%). The induced cells may differentiate into NK cells in vitro and show primary engraftment ability in bone marrow of NSG mice. Moreover, Tan et al. [68] also induced definitive HSPCs with engraftment abilities by transducing another transcription factor, MLL-AF4, into iPSC-derived HSPCs. Along with the assistance of SB431542, there was a remarkable increase in the percentage of CD34+CD45+ HSPCs (86.6%). These CD45+ cells showed a significant human cell chimerism (>20.0%) in the bone marrow, spleen, and peripheral blood of the NSG mice at 8 weeks after transplantation, showing the multilineage engraftment ability of the cells.
As shown above, the generation of definitive HSPCs has advanced by leaps and bounds. Despite this remarkable progress, there remains a long way to go for the optimal maturity and functionality of iPSC-derived HSPCs. One of the greatest limitations regarding these cells is their engraftment ability. Many attempts failed to generate engraftable HSPCs without teratoma formation or exogenous transcription factor expression [25]. Further investigation is needed to generate definitive HSPCs with full maturity and functionality, yet it cannot be denied that these trials have set a milestone for producing true HSPCs from iPSCs in the near future.
Applications of human iPSC-derived HSPCs
Human iPSC-derived definitive HSPCs, as shown from the above functional analysis, can be further induced into all types of hematopoietic cells, including myeloid (monocytes, macrophages, neutrophils, basophils, eosinophils, erythrocytes, megakaryocytes, and platelets) and lymphoid (B cells, T cells, and NK cells) lineages. Owing to these characteristics, iPSC-derived HSPCs may be broadly used for understanding the mechanisms of blood-related disorders and relevant therapeutic approaches. Primarily, the transplantation of differentiated HSPCs can be a therapeutic option for patients with hematologic malignancies to restore the impaired hematologic functions after chemotherapy. Furthermore, genetically modified iPSC-derived HSPCs can be utilized to establish models for deciphering the onset and progression of genetic hematologic disorders. By extension, a patient-specific drug screening platform can also be constructed with iPSC-derived HSPCs to develop personalized drugs. Accordingly, many researchers have recently reported various applications of iPSC-derived HSPCs in the field of regenerative medicine.
1. Genetic modifications of iPSCs: disease modeling and gene therapy for genetic blood disorders
The discovery of restriction endonucleases opened a new era in medical science. Restriction endonucleases are enzymes that bind and cleave DNA fragments at specific palindromic recognition sites, subsequently inducing DNA repair systems including non-homologous end joining and homology-directed repair (HDR) [69]. On the basis of these molecular scissors, customized artificial scissors, including zinc-finger nucleases, transcription activator-like effector nucleases, and the CRISPR/Cas9 system, were introduced, marking the development of genome editing technology [70]. Genome editing technologies may be used to modify iPSCs, which can be further differentiated into all hematopoietic lineages that may be used for disease modeling and gene therapy [71,72].
Numerous genetic blood disorders were modeled and treated via the replacement of malfunctioning blood cells with newly produced healthy blood cells based on iPSC technology and genome editing technology. For instance, hemoglobinopathy refers to a group of disorders caused by an abnormal production or structure of hemoglobin molecules [73]. SCD and thalassemia are 2 common types of hemoglobinopathy, and numerous efforts have been made to find their underlying mechanisms and ultimately cure these diseases. SCD is a hereditary disease resulting from a recessive mutation in the β-globin gene at the chromosomal region 11p15.5. A single nucleotide change causes the alteration of the codon of the sixth amino acid from glutamic acid to valine, which subsequently leads to abnormal hemoglobin, hemoglobin S (HbS) [74]. In 2020, Haro-Mora et al. [62] reported biallelic correction of a β-globin point mutation (HBB:c.20A>T) in iPSCs via HDR induced by CRISPR/Cas9 through electroporation. The corrected cells went through restriction fragment length polymorphism screening using the HhaI enzyme site (GCGC), and they were further induced into erythroid cells through an iPS-sac method. The generated cells contained normal definitive globin including the β-globin protein, implying that the further development of this method may be used as a future regenerative transfusion therapy for SCD. Aside from direct correction of the hemoglobin B (HbB) mutation, there has been an attempt to indirectly treat this disease by controlling the expression of certain genes to alleviate the symptoms. For example, high levels of fetal hemoglobin (HbF) are known to ameliorate SCD by mitigating its structural abnormalities. Therefore, B cell CLL/lymphoma 11A (BCL11A), a repressor of γ-globin expression and HbF production in adult erythrocytes, was targeted for the treatment of SCD [75]. Esrick et al. [76] reported post-transcriptional genetic silencing of BCL11A in CD34+ cells by shmiR-based gene knockdown. This allowed erythroid lineage-specific knockdown, which was validified by the successful engraftment of erythroid cells with stable HbF production ability (20.4%–41.3%) in patients, in which SCD manifestation decreased or disappeared. This approach underlines the possibility of SCD treatment by BCL11A downregulation at the iPSC level with higher efficiency and ease. This is due to the fact that gene modification t the iPSC level is advantageous in terms of screening for genetically modified cells, considering the difficulty of doing so in CD34+ cells. However, it has also been found that BCL11a deficiency causes the downregulation of cyclin-dependent kinase 6 and a subsequent delay in the cell cycle, which in turn results in HSPCs with defective self-renewal and multilineage differentiation, showing close resemblance to the aged HSPCs [77]. This limits the utilization of BCLL11A for SCD treatment by hindering its large-scale production, showing that further studies are in need for future therapeutic usage.
Meanwhile, thalassemia is another inherited disorder that is characterized by decreased hemoglobin production. It has been mainly classified into 2 different types, α- and β- thalassemia, where α-thalassemia is usually due to a deletion within the α-globin gene, while β-thalassemia is usually caused by non-deletional mutations [78]. The genetic correction of concurrent α- and β-thalassemia in patient-derived iPSCs by the application of the CRISPR/Cas9 system was presented by Li et al. in 2022 [79]. CRISPR/Cas9-mediated HDR with linearized donor DNA was conducted. The DNA was designed to target the heterozygous Hb-WS (ααWS/αα) mutation in the HBA2 gene and the homozygous β41-42 (TCCT) deletion in the HbB gene, which are common mutations for α- and β-thalassemia, respectively. The results showed the successful correction of thalassemia mutation in iPSCs, which retained normal pluripotency and multilineage potential to differentiate into erythroid cells. This shows that gene modification of iPSCs targeting thalassemia can be applied for autologous transplantation in patients with this disorder. As shown above, substantial progress has been made regarding genetic modifications of iPSCs to treat hematologic disorders. However, these products need further verification regarding their functionality, such as their ability to engraft. Nevertheless, there is no denying of the potential of iPSCs in the field of gene therapy.
2. iPSC in cellular immunotherapy: universal chimeric antigen receptor systems for hematologic malignancies
Chimeric antigen receptor (CAR) cell therapies, with CAR-T cells at the front line, have recently emerged as a major cellular immunotherapy for hematologic malignancies, including leukemia [80]. CAR cell therapy is a revolutionary cancer treatment method that utilizes CAR, an engineered synthetic receptor that recognizes and directs immune cells to eliminate a specific target antigen [81]. For instance, in 2020, 858 CAR-T cell therapies were under research or on the market worldwide, showing a 50% increase compared to the year before [82]. However, various limitations hinder the full utilization of the present CAR systems [83]. Currently, autologous CAR cells made with the same patient-derived immune cells are widely in use to avoid severe immune rejection caused by the mismatch of HLA between the donor and the recipient. Yet, due to the scarcity of patients’ healthy immune cells after repeated chemotherapy sessions, the collection of immune cells from patients’ peripheral blood mononuclear cells requires labor-intensive procedures, such as individual blood apheresis, making the entire process costly in terms of price and time. Therefore, “off-the-shelf” or universal CAR (UCAR) cell therapies have been suggested as a breakthrough to address the drawbacks of conventional CAR systems [84]. UCAR cell therapy utilizes allogenic immune cells from healthy donors, which are further fabricated using gene-editing techniques, such as disturbing the TCR gene and/or HLA class I loci of the allogenic T cells, to avoid complications such as GvHD [85]. Large-scale production of these UCAR cells is possible, thereby allowing improved quality and accessibility of the products.
Meanwhile, as the self-renewing ability of iPSC enables the indefinite production of cells and the availability of gene modification in iPSCs allows prefabrication of iPSCs according to one’s needs, iPSCs have emerged as a new source for UCAR cell production and thus have been intensely studied [84]. In 2022, several researchers reported the differentiation of human CAR+ iPSCs into highly functional CAR-T cells through 3D organoid culture methods [86,87]. The reported artificial thymic organoids allowed the selective differentiation of CD19-expressing CAR-iPSCs into fully functional CAR-T cells. Thus, in the same year, Wang et al. [88] reported the generation of hypoimmunogenic T cells from genetically engineered allogenic iPSC. Hypoimmunogenicity was established by the disruption of B2M, CITA, and NK cell-ligand receptor CD155 and the transduction of HLA-E, while CAR was expressed in iPSCs by engineering of CD20. These iPSC-derived T cells well escaped recognition by NKG2A+/DNAM-1+ NK cells and CD8/CD4+ T cells while maintaining the anti-tumor ability. Meanwhile, CAR-NK cells derived from iPSCs were also established by Li et al. in 2019 [89]. CAR containing factors (NKG2B transmembrane domain, 2B4 co-stimulatory domain, CD3y signaling domain) that mediate strong antigen-specific NK cell signaling, namely NK-CAR, were overexpressed in iPSCs through transfection of the PiggyBac transposon vector. NK-CAR expressing iPSCs were successfully differentiated into NK cells (NK-CAR-iPSC-NK cells), which showed prolonged survival and successfully inhibited tumor growth in an ovarian cancer xenograft model. Further studies are needed, but these trials suggest the potential of iPSC-induced CAR cells for anti-cancer immunotherapy.
3. Recapitulation of immune system within the organoid: a platform to mimic in vivo pathophysiological environment
Organoids are self-organizing 3D organotypic structures generated with either iPSCs, ASCs or tissue biopsies, which have recently been described as groundbreaking research platforms to simulate development, homeostasis, or pathological changes of the organs [12]. However, a lack of the microenvironment, including vasculature, stromal cells, and immune cells, is considered as a drawback of organoid systems [53,90]. Most of all, the immune system plays a crucial role in the pathogenesis of a wide variety of diseases, including infectious diseases, autoimmune diseases, degenerative diseases, and immune rejection. Thus, numerous studies have aimed to create an immune milieu within organoids to improve their pathophysiological relevance [17,91–93]. In this regard, iPSCs can be a valuable tool for embodying the immune system within organoids from the same source, as they have the potential to differentiate into not only parenchymal cells, but also hematopoietic lineages.
According to previous reports, 2 approaches have emerged to describe the concept of organoid-immune coculture: (1) a holistic approach and (2) a reductionist approach [94]. The holistic approach encompasses methods for developing immune cells innately within organoids during differentiation from iPSCs. For instance, Hwang et al. [95] validated the induction of primitive HSPC-like transcriptomes within iPSC-derived human kidney cortex organoids, considering that the kidney is one of major niches interacting with HSPCs. In addition, several researchers have established protocols for producing cerebral organoids containing microglia that are readily differentiated from mesodermal progenitors during organoid development [91,96]. In contrast, from the perspective of the “reductionist approach,” immune cells differentiated from iPSC-derived HSPCs are prepared separately from iPSC-derived parenchymal cells. Subsequently, these cells, by combining with other niche factors, are assembled in an optimized ratio and patterned into microstructures to mimic the architecture of the original organ. This strategy can be usefully employed for recapitulating the microenvironment with a high degree of cellular and structural complexity. For example, the intestinal epithelial barrier maintains homeostasis by interacting with neighboring stromal cells, the commensal microbiome, and mucosal immune system that is activated in response to intrusion of external pathogens or immunogenic components. As the imbalance among those components manifests as immunological disorders in the intestinal tract, reconstruction of the immune niche within the organoids is essential to closely mimic the pathogenesis of these diseases [94,97]. In this context, Tsuruta et al. [98] recently attempted to partially create an intestinal immune milieu by directly injecting iPSC-derived monocyte-like cells into the cavity of iPSC-derived gut organoids. In another implementation, cerebral organoids containing microglia populations obtained from iPSC-derived hematopoietic progenitor cells have also been produced with this reductionist approach [17,99]. Last but not least, iPSC-derived HSPCs can also be used to recapitulate the hematopoietic bone marrow, a microenvironment where homing, proliferation, and differentiation of HSCs take place. This establishment of the HSC niche within organoids has recently been demonstrated by Khan et al. [100]. They argued that bone marrow organoids mimicking the central bone marrow space could be generated with iPSCs that were embedded in mixed-matrix hydrogels. Through a series of differentiation processes, these organoids have been shown to retain the characteristic bone marrow components, including stroma, lumen-forming sinusoidal vessels, myeloid cells, and pro-platelets. Moreover, they successfully established a TGFβ-induced myelofibrosis model with these organoids and further utilized this system to screen for the efficacy of potential fibrosis inhibitors.
As shown so far, the development of iPSC-derived organoids interacting with environmental components, particularly immune cells differentiated from iPSCs of the same individual, may allow us to establish an advanced form of in vitro disease modeling. As this model closely captures disease pathogenesis and the relevant immune milieu, it can be a powerful tool to investigate the reciprocal crosstalk between immune cell populations and the corresponding organs. Moreover, this approach may eventually offer a promising personalized platform for drug discovery and contribute to the discovery of innovative therapeutics.
Conclusion
In this review, we presented an overview of the generation of HSPCs from iPSCs (Fig. 1). In vivo studies conducted using various animal models elucidated the entire developmental process of hematopoiesis, and thus many attempts were made based on those findings for the successful production of HSPCs in vitro. As these studies revealed that definitive hematopoiesis is the pathway that is capable of producing definitive HSPCs with multilineage potential, we in turn summarized selected reports of iPSC-induced definitive hematopoiesis. Hence, we reviewed the application of these iPSC-derived definitive HSPCs in terms of disease modeling and gene therapy through genetic modifications of iPSCs, cellular immunotherapy via a UCAR system, and pathological studies using organoids. Although further studies are needed to improve the maturity, functionality, and efficiency of iPSC-derived HSPCs, there is no doubt that these trials set a milestone for the ultimate production of definitive HSPCs in the near future.
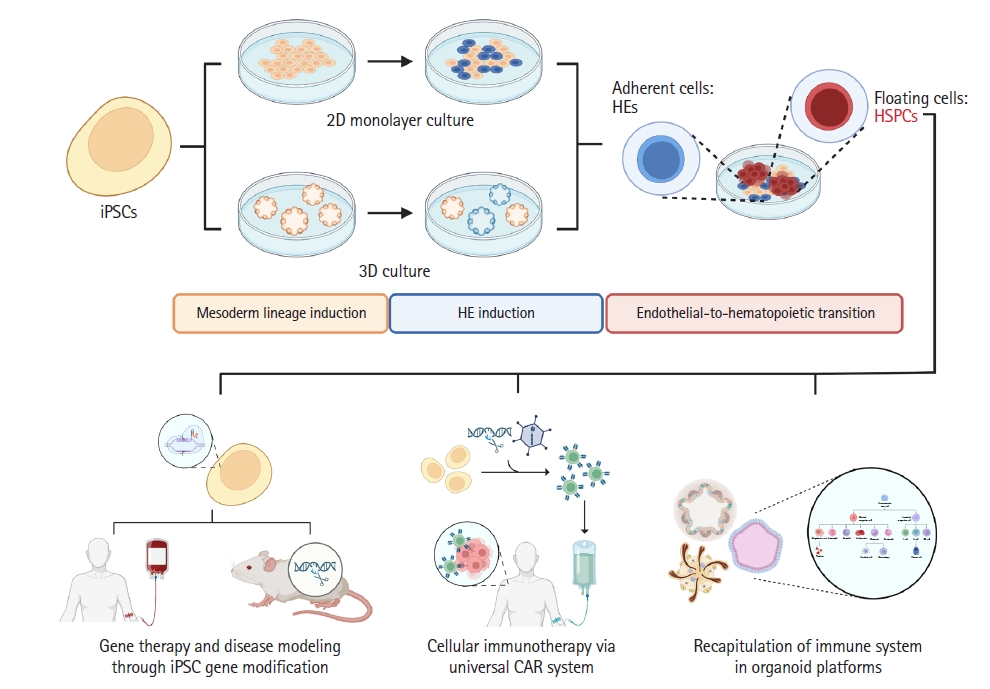
Graphical overview of the generation of hematopoietic stem and progenitor cells (HSPCs) from induced pluripotent stem cells (iPSCs). The upper part of the figure illustrates the production of definitive HSPCs from iPSCs in vitro. iPSC-derived definitive HSPCs may be generated through 2-dimensional (2D) monolayer or 3-dimensional culture (3D) methods, closely mimicking the process of definitive hematopoiesis, which consists of mesoderm lineage induction, hemogenic endothelium (HE) induction, and the endothelial-to-hematopoietic transition. The lower part of the figure gives an overview of the possible applications of iPSC-derived definitive HSPCs. These cells may be utilized in gene therapy and disease modeling after gene modification, may be used in cellular immunotherapy in the form of a universal chimeric antigen receptor (CAR) system, or may also recapitulate the immune system via iPSC-derived organoid platforms.
Notes
Conflict of interest
No potential conflict of interest relevant to this article was reported.
Funding
This research was supported by the Basic Research Program through the National Research Foundation of Korea (NRF) (2022R1C1C1009606) funded by the Korean government.
Data availability
Please contact the corresponding author for data availability.