Introduction
AlzheimerŌĆÖs disease (AD), the most common cause of dementia, is a worldwide public health concern that occurs in a meaningful proportion of elderly individuals [1]. The total number of deaths caused by AD has steadily increased as current therapies merely rely on alleviating symptoms, not on curing AD [1]. To develop a definitive cure for AD, scientists have poured their efforts into the discovery of novel pathogenic mechanisms leading to neuronal loss and deterioration of cognitive function in aging brains [2]. Transgenic mice have been widely employed to study the implications of several genes associated with familial AD (FAD) for AD progression [3]. For instance, transgenic mice with mutations in A╬▓ precursor protein (APP) and/or presenilin (PS) are gold-standard AD model systems as they recapitulate signature aspects of AD, such as neuroinflammation and the deposition of amyloid beta (A╬▓) and hyperphosphorylated-Tau (pTau) in the brain [3-7]. Despite these similarities, a number of discrepancies compared to human AD brains have been reported [8]. Although the accumulation of A╬▓ was observed, the formation of neurofibrillary tangles (NFTs) in neurons was not reproduced well at the same time in those animal models [8]. Moreover, the current transgenic mice models are only available to recapitulate FAD, not sporadic AD (SAD), which accounts for approximately 97% of AD cases [8]. To overcome the limitations of animal models having different genetic backgrounds from humans, humanized three-dimensional (3D) AD models, also known as AD organoids, have been suggested as alternatives to animal models [4,5,7,9-11].
AD organoids are 3D cell-cultured and self-organized AD models based on human cells or human induced pluripotent stem cells (iPSCs) expressing FAD or SAD-associated genes [4,5,7,9-12]. Various forms of AD organoids have been proposed that closely reproduce AD pathogenesis, including depositions of A╬▓ plaques and NFTs as well as promotion of neurodegenerative inflammation [4,6,7]. The organoids involve spheroids, 3D bioprinted brain tissues or vessels, brains-on-a-chip, and combined platforms [4,5,7,9-11]. The use of cell reprogramming systems, such as AD neurons derived from AD patient fibroblasts, allows reproduction of the genetic material of AD patients as well as AD pathology [10,13]. AD organoids enable dissection and reconstruction of extremely complex functions of healthy and/or AD human brains that are otherwise not possible in animal or human models [4,5,7,9-11]. Furthermore, these miniaturized models can allow several robust and high-throughput analyses under a controllable microenvironment. The microarchitectures of AD organoids are relevant and simple, and they are capable of recapitulating specific biology, mechanisms, and development of neurological disorders. Overall, AD organoids represent excellent models for understanding the progression of both FAD and SAD, which may contribute to the development of effective AD preventions and cures.
In this review, we explore details on AD organoids, in terms of (1) the major characteristics of AD to be implemented in AD organoids, (2) the design and preparation of various AD organoids, (3) advantages and potential challenges, and (4) future directions in this area.
Major characteristics found in AD
This section provides essential background regarding pivotal AD features to be replicated in AD models. The evaluation criteria are summarized in Table 1 [4-7,10,11,14-17].
1. Accumulation of A╬▓ aggregates
The accumulation of insoluble A╬▓ fibrils and plaques is the first checkpoint to determine the pathophysiological relevance of in vitro AD models [8]. The formation of insoluble amyloid fibrils and plaques from A╬▓ peptides has been considered as a primary characteristic found in AD patients at the early stages [18]. In normal conditions, APP is processed by ╬│-secretase, producing P3 fragments. In AD pathological conditions, APPs are truncated by ╬▓-secretase and ╬│-secretase, resulting in the production of various A╬▓ peptides depending on the cleavage site by ╬│-secretase. Although A╬▓1-40 (A╬▓40) peptides are the most abundant form, comprising 80%-90% of A╬▓ peptides, A╬▓1-42 (A╬▓42) peptides are neurotoxic as they are hydrophobic and prone to form aggregates, which further develop into oligomers, amyloid fibrils, and amyloid plaques, sequentially. In this regard, most in vitro AD models have utilized neuroprogenitor cells expressing FAD mutations, which are known to achieve higher A╬▓42/A╬▓40 ratios and the formation of A╬▓ plaques [8]. The most popular FAD mutations are APP mutations (e.g., KM670/671NL [Swedish], V717I [London], and V717F [Indiana]) and/or PS mutations (e.g., PS1-M146L, PS1-L166P, PS1-I213T, and PS2-N141I) [8]. In addition, SAD models with mutations in AD risk factors (e.g., APOE e4 or TREM2) also exhibited the robust formation of A╬▓ plaques [19,20]. Therefore, AD models can be evaluated by validating the presence of an increased soluble A╬▓42/A╬▓40 ratio and the accumulation of A╬▓ oligomers, fibrils, and plaques in the models [18].
2. Deposition of tau fibrils
The formation of NFTs in neuronal cells is the second hallmark to be validated upon completion of AD model preparation [8]. Tau is a microtubule-binding protein that stabilizes microtubules in normal physiological conditions [8]. During AD progression, tau proteins are hyperphosphorylated, detached from microtubules, and formed into paired helical filaments (PHFs), followed by NFTs, which are frequently found in AD models and patient biopsies [21]. Several studies also revealed the formation of NFTs was highly correlated with axonal degeneration, mitochondrial dysfunction, and synaptic dysfunction, which further increase neurodegeneration [21]. Increased kinase activity (e.g., GSK-3╬▓ and CDK5) is known as a modulating mechanism to induce the phosphorylation on multiple sites of tau (more than 7-8 phosphates per tau), resulting in pTau [22,23]. Other modifications, such as proteolytic cleavage generating a S258-I360 fragment and O-GlcNac glycosylation in the S/T-P motif, also promote the formation of tau aggregations in AD brains [24,25]. Growing evidence shows that A╬▓-driven increased glial activity produces proinflammatory factors that further increase tau accumulations in AD brains [4,6]. In addition, many AD patient brains exhibited aggregates of both A╬▓ and pTau, indicating the potential role of interactions between the two misfolded proteins in AD. Given the strong correlation between pTau accumulation and AD pathology, increased levels of pTau and the presence of PHFs and NFTs are standard markers to evaluate in AD models [18].
3. Neuroinflammation
The prominent activation of innate immune cells can be a promising marker to be monitored, as their phenotypes are closely correlated with the severity of AD [26]. Among the innate immune cells, astrocytes and microglia are two major components involved in the inflammatory responses to AD ques [4,12]. In the early stages of AD where protein A╬▓ and tau aggregates are starting to be formed in the brain, the polarization of anti-inflammatory astrocytes and microglia appears to be dominant. Astrocytes serve neuroprotective roles by secreting neurotransmitters supporting neuronal activity and removing toxins around neuronal cells [27]. Microglia also play neuroprotective roles by phagocytosing damaged synaptic parts, called synaptic pruning [12,28]. However, chronic exposure to AD conditions can alter the phenotype of both microglia and astrocytes, promoting proinflammation and neurodegeneration in AD brains [4]. In the later stages of AD, a number of studies demonstrated the induction of hyper-reactive astrocytes, promoting the significant levels of oxidative stress (hydrogen peroxide [H2O2] and nitric oxide [NO]) and the production of proinflammatory cytokines (interleukin [IL]-1╬▓, IL6, tumor necrosis factor [TNF]-╬▒, interferon [IFN]╬│, etc.) and chemokines (CCL1, CCL2, CXCL1, etc.) that further activate other proinflammatory components of the innate immune system [4,6]. Moreover, microglia infiltrate into regions with A╬▓ and NFT accumulations and display a detrimental phenotype that exacerbates AD pathology by increasing oxidative stress (superoxide [O2], NO), tau aggregates, and proinflammatory mediator levels (IL-1╬▓, IL-6, TNF-╬▒, etc.) [4]. In this regard, AD models can be evaluated by investigating the phenotype of innate immunity and assessing the increased soluble factors from innate immune cells.
4. Neurovascular dysfunction
Neurovascular dysfunction is a distinctive feature frequently observed in AD patient brains, which show an increased influx of neurotoxic blood-derived debris, microbial pathogens, and proinflammatory responses, all of which exacerbate neurodegeneration [29]. Neurovascular abnormalities have been strongly correlated with blood-brain barrier (BBB) disruption and attributed to genetic AD risk factors, A╬▓/tau pathology, and the degeneration of BBB components, such as endothelial cells and pericytes [29]. According to recent positron emission tomography and magnetic resonance imaging studies, BBB disruption was observed in approximately 40% to 80% of AD patients, particularly in the hippocampus region, a center for memory and learning processes [29,30]. Several studies have shown that AD patients possessing an AD risk factor such as APOE e4 have higher levels of cyclophilin A and matrix metalloproteinase 9 (MMP9) in the endothelium and pericytes of BBB [31,32]. Follow-up studies showed that the activation of cyclophilin A-MMP9 axis decreased BBB integrity [32,33]. Amyloid and tau depositions on the vascular wall can also decrease BBB integrity by inducing endothelial degeneration [34,35]. In addition, reduced pericyte numbers around the BBB and increased infiltrated-macrophages can increase BBB permeability [33,36]. BBB disruption can be confirmed by detecting any decrease in tight-junction markers (e.g., ZO-1, ╬▓-catenin, VE-cadherin, and claudin-5) [14]. The direct consequences of a leaky BBB can be evaluated by monitoring any increase in the permeability coefficient of fluorescent probes or a decrease in transendothelial electrical resistance across the BBB [7,15]. In addition, the consequences of BBB disruption can be markers of neurovascular dysfunction in AD; examples include any increased proinflammatory mediators, accumulated blood-derived proteins (e.g., albumin, plasminogen, fibrinogen, and immunoglobulin G), and deposited neurotoxic debris or microbial pathogens [29].
5. Neurodegeneration
A significant reduction in brain mass is a major hallmark of late-stage AD, leading to clinical symptoms such as progressive decline in memory, thinking, language, and essential body functions [1]. Since the assessment of clinical symptoms is impossible within in vitro models, the detection of neuronal damage and neuronal loss is an alternative criterion to be validated in AD models. The induction of synaptic impairment followed by the loss of neural function has been observed in both AD patients and in vitro AD models [4,37,38]. Evidence of synaptic impairment involves reductions in major synaptic proteins (e.g., calsyntenin, GluR, neurexin, syntaxin, and synapsin) that participate in core synaptic processes such as dendritic spine assembly, postsynaptic Ca2+ signaling, synaptic transmission, synaptogenesis, and presynaptic differentiation [39]. Decreased neural activity can be assessed by the neural responses toward optical, electrical, or chemical stimulation. For instance, several studies showed reduced intracellular Ca2+ levels in AD neural cells in response to an action potential buffer [4]. The most apparent evidence of neurodegeneration is the reduction of neural population in AD models. To this end, a number of studies validated neurodegeneration by showing a decrease in neuronal markers or the viability of AD models [4,6,7].
AD spheroids
Multicellular spheroids represent a simple form of organoids used to predict pharmacokinetics and the therapeutic efficacy of candidate drugs in the brain [10,11,14]. A spherical cluster of neural cells retains a 3D configuration and closely mimics the functional and structural properties of the brain tissue [10,11]. In addition, BBB-spheroids can be prepared by wrapping the brain spheroid with an additional layer of brain endothelial cells that recapitulate the penetration of medicines or pathogens across the BBB [14]. Therefore, spheroids serve as an excellent brain disease model for the preclinical evaluation of therapeutics targeting neurological disorders, including AD.
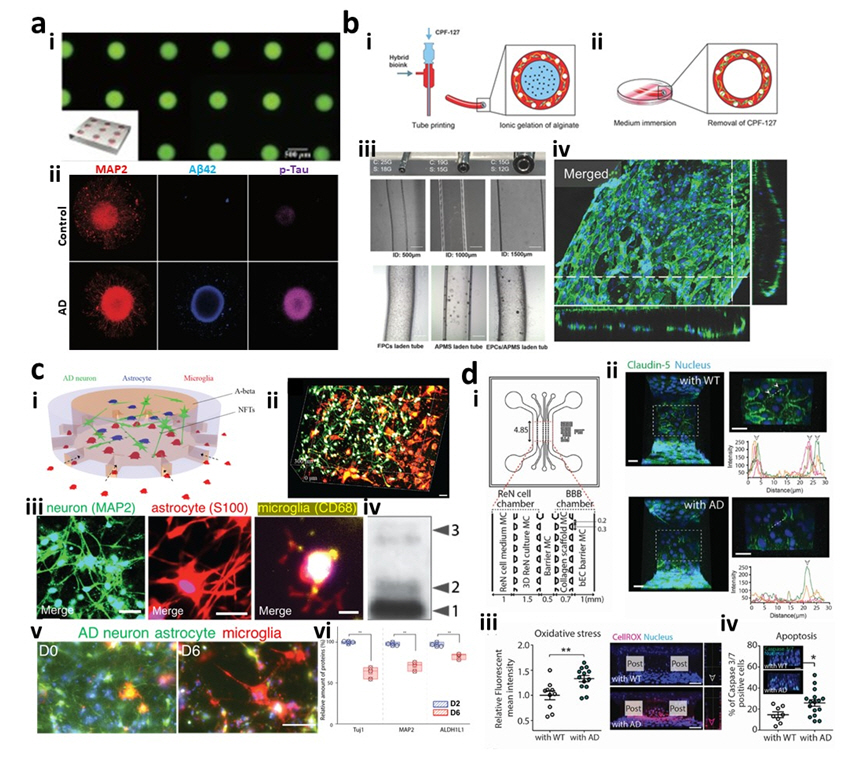
A method to create AD spheroids is to culture AD neuronal cells on non-cell adherent surfaces or hydrogels not containing integrin-binding motifs (e.g., agarose, polyethylene glycol, or F127-DA hydrogels), which facilitate cell-cell interactions leading to the formation of cell aggregates [40-42]. Lee et al. [10] cultured AD patient-derived neural cells on ultra-low-attachment plastic plates and generated 3D human AD neurospheroids. The resultant AD patient neurospheroids displayed AD signatures such as the production of A╬▓40 and A╬▓42 and deposition of pTau. These personalized AD neurospheroids in an array format were used to screen various AD treatments including BACE1 and ╬│-secretase inhibitors in a high-throughput manner. However, the neurospheroids prepared by this method were not homogeneous in size and shape. To precisely control the size and shape of spheroids to achieve more consistent results, a number of studies employed a hanging drop or a micro-mold method [40,42]. In the hanging drop method, cells are in the culture medium suspended to the underside of petri dish lids [43]. Under the force of gravity, cells are congregated onto the bottom part of the hanging drop and form spheroids. The size of spheroids can be adjusted by changing the cell seeding density. Nzou et al. [14] showed a human cortex spheroid mimicking a functional BBB prepared by hanging drop culture. The resultant BBB spheroid was employed to test adverse effects of inorganic mercury on the human cortex region. The micro-mold method can also be used to determine the size and shape of spheroids by adjusting cell culture mold design [40,42]. Jorfi et al. [11] cultured human neural progenitor cells (ReN) overexpressing GFP (control) or APP/PS1-GFP (AD) in uniformly sized polydimethylsiloxane (PDMS) microwells (Fig. 1A). This study achieved notable neurite outgrowth from the spheroids. In addition, AD spheroids exhibited higher expression levels of A╬▓42 and pTau than control spheroids.
Taken together, AD spheroids are advantageous as the preparation methods are simple and readily accessible for most labs, enabling efficient mass production. The major disadvantage of spheroids is the lack of diffusion of nutrients and oxygens in the core if their size exceeds 500 ┬Ąm, leading to the formation of a necrotic core [44]. To solve this limitation, recent studies introduced perfusable vascular units into the spheroids, which supplied nutrients and oxygen to the deeper parts of spheroids [45].
3D-bioprinted AD brains
The human brain is the most complex organ in the body over a thousand sub-regions, serving distinctive functions. Multiple components relevant for remodeling human brains, such as the cell population, density, orientation, and microenvironments, vary depending on the region. In the context of complexity, 3D bioprinting is a promising technique to construct AD brain models, particularly those associated with BBB, as it allows the layer-by-layer deposition of cell-laden bioink with different compositions [9].
Inkjet bioprinting is a technique to pattern multiple droplets of low-viscous and soft bioink on the substrate [46]. Droplet formation can be controlled by a piezoelectric or electrostatic actuator, which applies impulsive voltage causing a pressure pulse on the nozzle orifice. The instantaneous volume increase of bioink caused by the pressure pulse results in the ejection of bioink from the nozzle and its deposition on the substrate. For instance, Lorber et al. [47] successfully printed adult rat ganglia and glial cells using a piezoelectric inkjet printer. Thermal actuators are another system used to control the formation of cell-laden droplets on the substrate by creating heat pulses, inducing partial vaporization, and generating pressure pulses for the ejection of bioink. Xu et al. [48] employed thermal inkjet printing to create a 3D neural sheet composed of primary neural cells from rat hippocampal and cortical tissues and fibrin gels. However, several studies reported that the frequencies (15-25 kHz) of electric pulses and the local temperatures (~300┬░C) of heat pulses could reduce the viability of mammalian cells during printing, which may limit the application of inkjet printing to create functional 3D brain organoids. Recently, Sharma et al. [49] patterned 3D neural tissue with neuroprogenitor cells derived from iPSCs on the substrate under mild conditions using a microfluidic-based inkjet platform. Instead of using thermal or piezoelectric control, this system applied specific pressures to each channel to control the crosslinking and printing rate. Cell-laden bioink (a mixture of fibrinogen, alginate, and genipin) was mixed with a crosslinker at the junction of bioink and crosslinker channels to initiate gel solidification at the printhead of microfluidics. The cell-laden hydrogels were then transferred onto the substrate and formed into an array of semi-spherical neural tissue. Inkjet bioprinting allows high-resolution cell placement in the pico-liter range; however, the use of low-viscosity bioink limits the ability to pile up multiple layers of bioink in the Z-direction.
Extrusion-based bioprinting is a 3D bioprinting technique with viscous bioink that deposits a continuous filament and builds multiple layers. Gu et al. [50] generated a cubic-shaped neural mini-tissue by microextrusion bioprinting of human neural stem cells laden with bioink including the polysaccharides alginate, agarose, and carboxymethyl chitosan. The stiffness of the resultant mini-tissue (0.8-7.5 kPa) was comparable to that of human brains, as well as in vivo models. In addition, the printed neural stem cells were differentiated into GABAergic neurons, astrocytes, and oligodendrocytes. Miller et al. [51] printed a 3D filament of carbohydrate glass surrounded by a hydrogel encapsulating mouse embryonic cells. The filament is a sacrificial template creating a lumen area so that human umbilical vein endothelial cells were added and formed into vascular structures surrounded by tissue. Furthermore, the use of a coaxial nozzle composed of an outer and inner needle can extrude a two-layered filament with the inner layer to be removed later so that enable to create vascular structures in a single step [52]. Gao et al. [53] utilized coaxial extrusion to generate a blood vessel structure with endothelial progenitor cells (EPCs) covered by the vascular-tissue derived decellularized extracellular matrix (VdECM) (Fig. 1B). The hybrid bioink with EPCs and VdECM was loaded into the shell nozzle while Pluronic F-127 was in the core nozzle. Upon the completion of extrusion, the outer layer was solidified by thermal gelation and the inner layer was chemically removed by CaCl2 solution. In this study, the resultant blood vessel along with a proangiogenic drug (atorvastatin) was successfully implemented in the ischemic limbs of mice and contributed to limb salvage. The major drawback of extrusion is the low speed of building up 3D structures and its limitation to viscous bioink, unlike other bioprinting methods; however, extrusion would be advantageous for constructing continuous multiple-layered vascular structures in a single step [9]. With improvements of the mechanical properties of hydrogel bioink, bioprinted models could be further employed not only for in vitro drug screening, but also for vascular replacements [53].
Overall, the 3D bioprinting technique can generate personalized 3D neural tissues or BBB constructs by stacking either cell-laden building blocks or extruded filaments on the substrate. It should be noted that there is no standard AD model prepared by 3D bioprinting as the current bioprinting technique is still in the early stages of development. Nonetheless, there is tremendous potential for improvement to create novel 3D models of AD brain tissues by 3D bioprinting in the future.
AD brains-on-chips
Neural cell culture in microfluidic devices, also referred as brains-on-chips, have received extensive attention for their capability to miniaturize human brains. In this platform, multiple cells can be cultured within district compartments connected through networks of channels [4,7,17,54]. Therefore, this organoid enables the compartmentalization of each brain region, thereby simplifying the extremely complex functions of healthy and/or diseased human brains. Since the miniaturized devices are very thin and optically transparent, other optical, optochemical, and electrical measurement techniques have been integrated in microfluidics. Therefore, chip-based AD models allow multiple robust and high-throughput analyses in parallel.
Microfluidics has emerged as a single-cell culture platform that closely mimics the key cellular activities of specific cell types contributing to AD pathology [4,7]. In addition, microfluidic-based cell culture is advantageous in terms of the liquid handling system, which enables not only continuous supply/removal of nutrients and waste, but also the generation of stable chemical gradients for single-cell chemotaxis. Cho et al. [54] developed a chemotactic microfluidic platform that allowed the separation of stimulated human microglia subpopulations in vitro according to the use of soluble A╬▓ or insoluble A╬▓, extracted from AD brain tissues. In addition, they further created an in vitro 3D cylindrical capillary-like structured BBB integrated with chemotactic channels, allowing a robust screening of BBB-targeting drugs targeting neurological disorders including AD [17]. This single-cultured microfluidic platform enabled the dissection and reconstruction of specific cellular responses toward AD ques; however, the single-culture models did not consider multicellular crosstalk in AD pathology.
The advances in cell culture within microfluidics enable the construction of multicellular systems and increase the feasibility of AD models. Park et al. [4] engineered a 3D human AD brain model along with innate immunity by co-culturing human neuroprogenitor cells expressing APP/PS1 and adult microglial cells in a 3D microfluidic platform (Fig. 1C). In this model, two compartments for APP/PS1 neuroprogenitor cells and microglia were linked by multiple channels, allowing microglial migration in response to soluble factors from the AD chamber with neuroprogenitor cells. This AD model provided key representative AD features: pathological accumulation of A╬▓ and pTau, and microglial proinflammation leading to damage of AD neurons. This model was further evolved with the use of iPSC-microglia to explore the pivotal roles of TREM2 in the migration and phagocytic activities in response to soluble A╬▓, A╬▓ plaques, and 3D cultured AD brain models. Shin et al. [7] combined a 3D AD model with APP/PS1 neuroprogenitor cells and a 3D tubular BBB with hCEMC/D3 in a microfluidics platform to explore the impact of BBB dysfunction in AD pathology (Fig. 1D). Two parallel channels (an AD compartment and a BBB compartment) were connected by additional chambers with collagens allowing crosstalk between cells in the AD and BBB compartments. This study revealed that the increased production of A╬▓, MMP2, and IFN╬│ in the 3D AD models contributed to the induction of reactive oxygen species and reduction of tight junctions in 3D BBB. This in vitro model validated the major signature of AD, including an increase in BBB permeability leading to an increased influx of neurotoxins through the impaired BBB, followed by neuronal damage. In this regard, multicellular AD models in microfluidics enabled the integration of multiple functions in a single platform, which significantly improved both the physiological relevance and the efficacy of analyses in comparison to other in vitro models.
Taken together, the use of microfluidics allows the precise modulation and simplification of intricate biological parameters in the study of AD brains [4,5,7,9-11,55]. In addition, microfluidic-based AD models have been combined with other analytic tools to provide tractable and quantifiable platforms for the study of AD pathology in both qualitative and quantitative manners. Therefore, chip-based AD models represent excellent platforms for the study of extremely complex functions of AD human brains that were otherwise not possible in animal or human models. A major challenge of cell culture in microfluidics is the use of PDMS, which absorbs significant amount of nutrients and testing molecules, leading to dilution of components in the culture medium [56]. Another concern is the maintenance of physiologically relevant pH and oxygen levels inside microfluidics due to the use of minute volumes of media for the cell culture [56]. To this end, the culture medium should be replaced routinely, which can be automated by a perfusion system [57]. Even if the preparation of microfluidics requires complex and specialized equipment that is not usually affordable for most cell culture laboratories, enormous efforts have been invested in achieving lower-cost and more standardized systems for microfluidic-based 3D cell culture.
Outlook
AD is a progressive and irreversible brain disease threatening the lives of millions of people throughout the world [1]. However, most recently-developed treatments for AD have failed to show any therapeutic efficacy in clinical phases even if they achieved impressive outcomes in preclinical trials with animal models [2]. In this review, we highlighted major human brain organoids for the study of AD pathology and the discovery of promising therapeutics for AD. Recently, human AD brain organoids have emerged as alternatives as promising preclinical models since they can precisely recapitulate the architecture and functionality of human AD brains [4,5,7,9-11]. In addition, brain organoids provide tractable cellular platforms in combination with other analytical techniques that enable robust and accurate screening of drug candidates for AD in a controllable manner. The utilization of AD patient-driven iPSCs further improves physio-pathological features, both for FAD and SAD [10,13]. Brain organoids have recently been combined with other organs to form body organoids for testing the systemic effects of drug candidates [58,59]. Given the rapid progress of the development of AD organoids, we believe that they will offer novel experimental platforms to understand the nature of brain involvement in AD pathology and to accelerate the discovery of novel cures for AD in the near future.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print