Bronchioalveolar organoids as a tool to study transforming growth factor-β and cigarette smoke–induced lung pathology
Article information

Abstract
Background
Respiratory medicine has high barriers to new drug development, with fewer approved new treatments and candidate drugs and a higher failure rate than other common disease fields. Most of the major candidate drugs identified in preclinical animal studies fail in the clinical setting because of differences between animal models and humans. Therefore, the rapid development of 3-dimensional (3D) organoid-based disease models that recapitulate human pathological development has attracted increasing attention in drug development and personalized medicine.
Methods
In the present study, we generated bronchoalveolar organoids (BAOs) from human pluripotent stem cells (hPSCs) and assessed their potential as a pulmonary disease model.
Results
Derived BAOs contained the expected spectrum of differentiated cells, including alveolar progenitors, type 1 and 2 alveolar epithelial cells, basal cells, secretory cells, ciliated cells, and mesenchymal cells. When the BAOs were exposed to transforming growth factor-beta, both fibrosis- and inflammation-related transcripts were significantly upregulated compared to the control. In addition, the exposure of BAOs to cigarette smoking extract induced increased levels of nitric oxide in a dose-dependent manner, as well as upregulating oxidative stress-related and pro-inflammatory genes.
Conclusion
These findings suggest that hPSC-derived BAOs could be a promising platform for modeling pulmonary fibrosis and chronic obstructive pulmonary disease and testing drug efficacy.
Introduction
Chronic lung disease slowly damages the lungs and causes a decline in lung function. Two of the most common chronic lung conditions are pulmonary fibrosis (PF) and chronic obstructive pulmonary disease (COPD), which are serious, lifelong lung diseases that make breathing difficult. PF and COPD share many symptoms, but can damage the lungs through different mechanisms.
PF is a rare but chronic, irreversible lung disease with a median survival time of 3 to 5 years after diagnosis. The treatment of PF is limited to 2 Food and Drug Administration-approved drugs, pirfenidone and nintedanib, which only slow the disease progression and reduce mortality and do not restore lung function [1,2]. PF is characterized by the accumulation of myofibroblasts and the excessive deposition of extracellular matrix (ECM) elements, which lead to failure of lung function. Although the cause of PF has not yet been identified, most risk factors involve a fibrotic mechanism through the activation of the transforming growth factor-beta (TGF-β) signaling pathway [3,4]. COPD, another respiratory disease, causes persistent and progressive respiratory symptoms, including shortness of breath, coughing, and/or phlegm production, after onset [5]. Cigarette smoking is the most common risk factor for COPD [6,7]. Cigarette smoking causes emphysema, which refers to the destruction of small air sacs at the ends of the airways of the lungs [8]. Furthermore, the airways become inflamed, which not only leads to chronic bronchitis but also adversely affects the bronchial epithelial cell repair process [9]. An important mechanism in the pathogenesis of COPD is oxidative stress, which amplifies chronic inflammation, fibrosis, and emphysema [10]. Thus, antioxidative therapies such as the administration of multiple antioxidants or boosting the endogenous levels of antioxidants have been developed to treat COPD [11].
Currently, multiple studies are underway to identify the pathogenesis of respiratory diseases such as PF and COPD and develop new drugs using in vitro human lung epithelial and fibroblast cell lines and in vivo animal models [12–14]. However, cell lines respond to external stimuli differently and exhibit altered phenotypic and functional properties compared to primary human lung cells. Animal models are also limited in their ability to recapitulate human PF and COPD progression and responses to therapy. Recently, 3-dimensional (3D) tissue models derived from human pluripotent stem cells (hPSCs) have emerged as promising respiratory disease models that can mimic human lung structure, function, and cell-matrix interactions better than cell line-based 2-dimensional (2D) culture condition models [15,16]. Three-dimensional organoids derived from hPSCs are suitable models for studying basic cell-cell and cell-matrix interactions, physiologically mimicking the organization and microenvironment of fibrotic tissue [12,17,18]. Here, we generated 3D bronchoalveolar organoids (BAOs) from human hPSCs and assessed their potential for modeling respiratory diseases. Our results suggest that hPSC-derived BAOs could be a promising platform for modeling PF and COPD and testing drug efficacy.
Materials and Methods
Ethics statement: Ethical approval for this study was obtained from the Public Institutional Review Board Designated by Ministry of Health and Welfare, Korea (P01-201411-ES-01).
1. Maintenance of hPSCs
Human PSCs (CHA15 and iPS-NT4-S1) were kindly provided by CHA University, South Korea. Briefly, the cells were cultured under xeno- and feeder-free conditions using TeSR-E8 (#05990; STEMCELL Technologies, Vancouver, BC, Canada) on dishes coated with Vitronectin XF (#07180; STEMCELL Technologies). Cells were subcultured at 80% confluency and passaged every 4 to 5 days by mechanical dissociation. All cells were incubated at 37°C in 5% CO2.
2. Stepwise differentiation of hPSCs into BAOs
Stepwise direct BAO differentiation was performed as previously described [19]. Briefly, undifferentiated hPSCs (unhPSCs) were plated in dishes coated with Matrigel matrix (Corning Life Sciences, Corning, NY, USA). unhPSCs were prepared at a density of <7 to 8 colonies per well. After overnight incubation, BAO differentiation was initiated with exposure to stepwise induction medium. UnhPSCs were incubated in step 1 medium (RPMI1640 [Gibco, Carlsbad, CA, USA] medium for 1 day. On the second day, the cells were incubated with step 1 medium containing 0.2% fetal bovine serum (FBS; step 2 medium) for 1 day. Then, colonies were cultured with step 2 medium containing 2% FBS for 2 days to induce definitive endoderm (DE). DE cells were incubated with step 3 medium (DMEM/F12 [Gibco] medium supplemented with 1X B-27 supplement, 1% penistrep solution [Sigma-Aldrich Corporation, St. Louis, MO, USA], 10 mM HEPES [Sigma-Aldrich Corporation] buffer and 2 mM L-glutamine) containing cytokines (200 ng/mL Noggin, 1 μM SAG [Sigma-Aldrich Corporation], 500 ng/mL fibroblast growth factor 4 [FGF4], 2 μM CHIR99021 [Stem Cell Technologies, Vancouver, Canada], and 10 μM SB-431542 [R&D Systems, Abingdon, United Kingdom]) for 5 days (day 4 to 9). Anterior foregut (AF) spheroids were harvested at day 9 of differentiation from those that floated naturally on the medium, and the aggregated spheroids were also physically separated from DE colonies. Finally, the AF spheroids were cultured with step 3 medium supplemented with 1% FBS and 500 ng/mL FGF10 (R&D Systems) (since day 10) to induce BAOs. The medium was changed every 48 hours. Derived BAOs were characterized at day 35 of differentiation.
3. Preparation of cigarette smoke extract
Research-grade cigarettes (3R4F) were obtained from the Kentucky Tobacco Research Development Center (The Tobacco Research Institute, University of Kentucky, Lexington, KY, USA). Using one 3R4F cigarette, cigarette smoke was bubbled through 10 mL of serum-free DMEM/F12 supplemented with 1% penicillin-streptomycin with a peristaltic vacuum pump for 1-2 minutes. Next, the solution containing smoke was filtered through a 0.22-μm filter to remove large particles and was regarded as a 10% cigarette smoke extract (CSE) solution. The CSE was prepared within 30 minutes.
4. Nitric oxide production assay
Nitric oxide (NO) production was detected by the modified Griess method. The Griess reagent (modified) (#G4410) was purchased from Sigma (St. Louis, MO, USA), and 250 mL of water was mixed with stock to make 1X Griess reagent. Briefly, hPSC-BAOs were cultured in a 6-well plate and treated with different concentrations (0% to 4%) of CSE for 8 days. The culture supernatants were collected and mixed with an equal volume of 1X Griess reagent in a 96-well plate at room temperature (RT) in the dark. The absorbance at 540 nm was measured with a microplate reader within 15 minutes (BioTek Inc., Shoreline, WA, USA).
5. RNA isolation and quantitative real-time polymerase chain reaction
Total RNA was isolated from hPSC-derived BAOs using an RNeasy Mini kit (#74004; Qiagen, Hilden, Germany), and cDNA was synthesized using TOPscrip RT DryMIX (#RT200; Enzynomics, Daejeon, Korea). Polymerase chain reaction (PCR) amplification was performed using a Step One Plus real-time PCR system (Applied Biosystems, Warrington, UK) with TOPreal qPCR 2X PreMIX (Enzynomics). All the mRNA expression data were normalized to GAPDH. The primer sequences for human genes are listed in Table 1.

Human primer sequences used for quantitative polymerase chain reaction
6. Immunohistochemistry and hematoxylin and eosin staining for HistoGel
The BAOs were embedded in HistoGel (Fisher Scientific, Waltham, MA, USA), sectioned at a thickness of 5 μm, and mounted on slide glass. The slides were rinsed in xylene and with decreasing concentrations (100% to 80%) of ethanol for deparaffinization and rehydration. The immunohistochemistry slides were subjected to hydrated autoclaving using an automated antigen-retrieval with a citrate buffer (0.01 M sodium citrate, pH 6) at 121°C for 1 minute and cooled for 30 minutes before immunostaining. They were then treated with Dako REAL peroxidase-blocking solution (Agilent, Santa Clara, CA, USA) for 20 minutes at RT and washed with cold phosphate-buffered saline with 0.1% Tween 20 (PBST) for 10 minutes at RT. The slides were blocked with 10% normal goat serum for 1 hour at RT and incubated with primary antibodies (NK2 homeobox 1 [NKX2.1], epithelial cell adhesion molecule [EPCAM], mucin 5A [MUC5A], P63, secretoglobin family 3A member 2 [SCGB3A2], platelet-derived growth factor receptor beta [PDGFRβ], Clara cell 10-kDa protein [CC10], SPC, T1α and aquaporin-5 [AQP5]) in blocking buffer overnight at 4°C. The next day, goat anti-mouse IgG (H+L) cross-adsorbed secondary antibody, Alexa Fluor 488 (A11001; Invitrogen, Waltham, MA, USA) and Alexa Fluor 647 (A28181; Invitrogen) secondary antibody were applied for 1 hour at RT in the dark. Eventually, the slides were washed with PBST (0.1% tween) and covered with Fluoroshield mounting medium with 4', 6-diamidino-2-phenylindole (DAPI). Additionally, hematoxylin and eosin (H&E) section slides were stained for the nuclei using hematoxylin (BBC Biochemical, McKinney, TX, USA) and rinsed with water, followed by a short exposure of acidic ethanol. The slides also were exposed to eosin, targeting the cytoplasm, and dehydrated through a sequential concentration change, from 70% ethanol to 100% ethanol. Section slides were mounted with Permount mounting medium (Fisher Scientific) after 3 exposures to xylene.
7. Statistical analysis
The statistical analysis was executed using GraphPad Prism 5 (GraphPad Inc., La Jolla, CA, USA). Values for all measurements are presented as means±standard deviation. The Student t-test was used for comparisons between 2 groups. All tests utilized one-sided methodology. A p-value of less than 0.05 was considered statistically significant.
Results
1. Generation and characterization of BAOs from hPSCs
Based on our robust feeder-free AF endoderm (AFE) differentiation method, we harvested AFE spheroids between days 9 and 12 of differentiation and cultured them for 40 days in BAO induction medium. The 3D morphology of the BAOs was evaluated histologically by H&E staining of cut slices from HistoGel-embedded blocks. The BAOs derived from hPSCs contained airway-like and alveolar-like cells (Fig. 1A). Immunofluorescence staining also showed that hPSC-BAOs expressed markers of alveolar progenitors (NKX2.1), distal/alveolar epithelial cells (EpCAM), goblet cells (MUC5A), basal cells (P63), secretory cells (SCGB3A2), mesenchymal stromal cells (PDGFRβ), club cells (CC10), type 1 alveolar epithelial cells (AEC1, T1α and AQP5), and type 2 alveolar epithelial cells (AEC2, surfactant protein C [SFTPC]) (Fig. 2A). Moreover, these markers were more strongly expressed in hPSC-BAOs than in unhPSCs cultures (Fig. 2B). These results demonstrate that the BAOs differentiated from hPSCs were composed of diverse cell types of alveolar and bronchiole tissues.
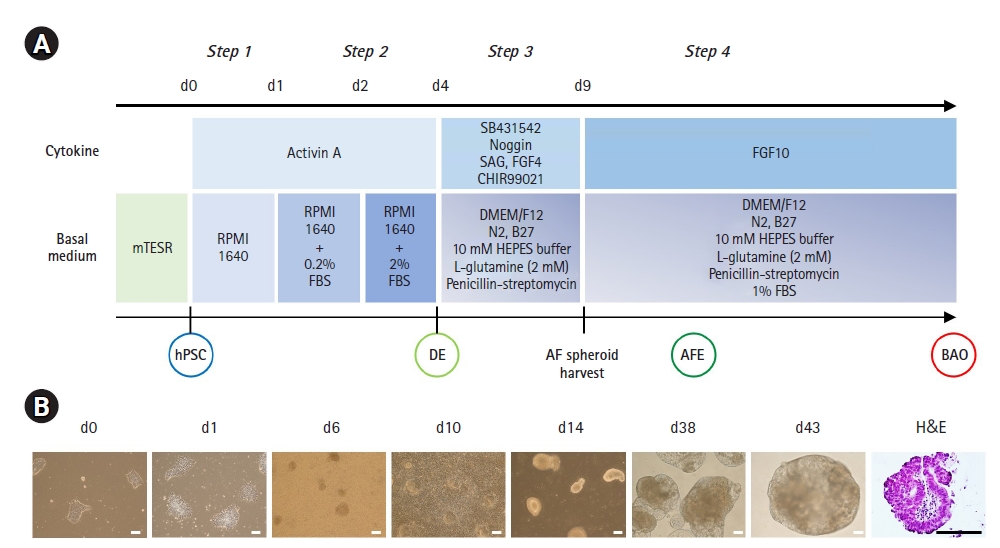
Generation of bronchoalveolar organoids (BAOs) from human pluripotent stem cells (hPSCs). (A) A schematic overview of the stepwise BAO induction protocol for human embryonic stem cells (hESCs) (CHA15) and human induced PSCs (hiPSCs) (iPS-NT4-S1). (B) Representative bright-field images and hematoxylin and eosin (H&E) staining image of cells and spheroids for days 0 to 43. Scale bars, 100 μm (bright-field images) and 200 μm (H&E staining). FGF, fibroblast growth factor; FBS, fetal bovine serum; DE, definitive endoderm; AF, anterior foregut; AFE, AF endoderm.
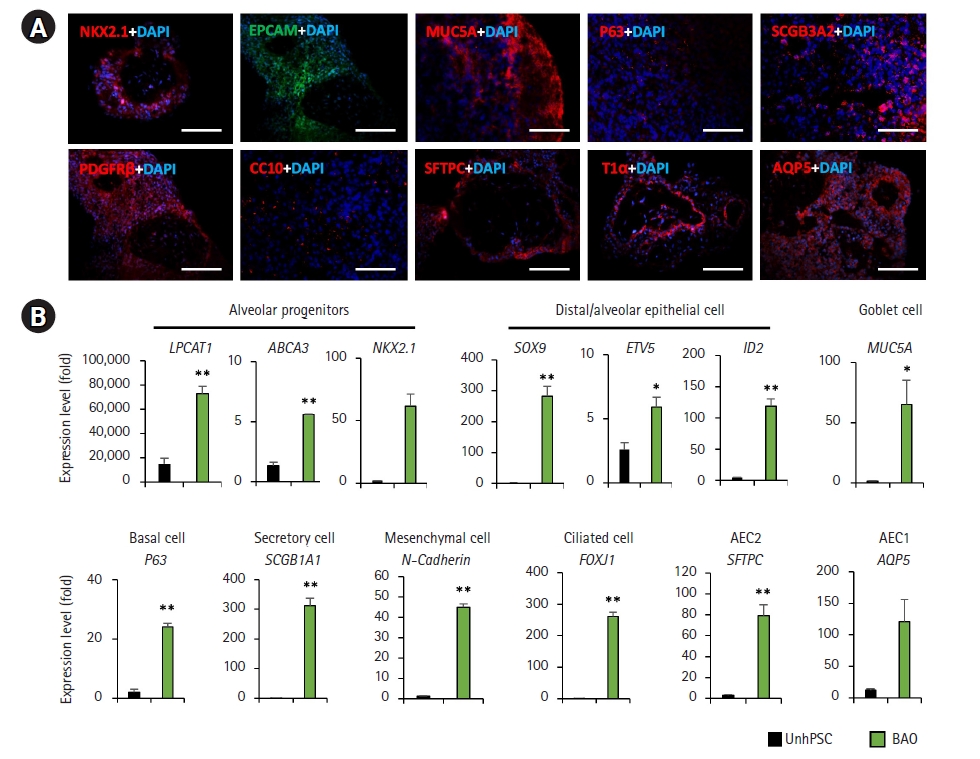
Characterization of human pluripotent stem cell (hPSC)-bronchoalveolar organoids (BAOs). (A) Representative images of immunostaining alveolar progenitor markers, distal/alveolar epithelial markers, alveolar epithelial cell (AEC) type 1 markers (T1α and aquaporin-5 [AQP5]), AEC type 2 markers (surfactant protein C [SFTPC]), and markers of other bronchioalveolar cells (goblet, basal, secretory, mesenchyme, and club cells) in the hPSC-BAOs. Scale bars, 200 μm. (B) Quantitative polymerase chain reaction of the markers of indicated alveolar progenitor cells, distal/AECs, AEC type 1 and 2 cells, and other bronchioalveolar cells (goblet, basal, secretory, mesenchyme, and multiciliated cells) in hPSC-BAOs. The data are shown as fold-change relative to undifferentiated hPSCs (unhPSCs). The bars indicate the mean±standard deviation. *p<0.05, **p<0.01.
2. TGF-β1 treatment induced fibrotic changes in hPSC-BAOs
hPSC-BAOs are predicted to be a useful model for evaluating lung fibrotic and inflammatory diseases among lung diseases. TGF-β1 is a major profibrotic cytokine that causes fibrotic changes via modulation of fibroblast phenotype and function, inducing myofibroblast transdifferentiation. Focusing on TGF-β1-mediated lung fibrosis, we treated day 47 hPSC-BAOs with TGF-β1 (25 ng/mL) for 72 hours and investigated the inflammatory and fibrotic responses at the transcriptional level. The expression levels of TGF-β1-mediated ECM-related (collagen type 1 alpha 1 [COL1A1]), myofibroblasts (alpha-smooth muscle actin [α-SMA]) and fibrotic genes (matrix metallopeptidase 9 [MMP9]), as well as pro-inflammatory cytokines (interleukin 1-beta [IL-1β], IL-6, and IL-11), were significantly increased after TGF-β1 treatment (Fig. 3A). These findings indicate that hPSC-BAOs responded to TGF-β1-mediated fibrotic stimulation, recapitulated lung fibrosis, and induced additional inflammatory responses.

Induction of transforming growth factor-beta (TGF-β1)-mediated fibrosis and inflammation in human pluripotent stem cell (hPSC)-bronchoalveolar organoids (BAOs). Expression of fibrosis and inflammation-related genes in TGF-β1-induced fibrotic hPSC-BAOs. The bars indicate the mean±standard deviation. **p<0.01.
3. CSE exposure induced inflammation and oxidative stress in hPSC-BAOs
CSE exposure has been established as a principal contributor to COPD. Toxic substances from CSE penetrate into the bronchial epithelium and directly affect bronchioalveolar cells. The toxic substances sustain the inflammatory response through the activation of immune cells and the release of pro-inflammatory mediators. In addition, NO regulates the tone of blood vessels and bronchoalveolar cells in the immune system, but their breakdown causes tissue damage due to oxidative stress. We analyzed the concentration of NO generated by exposure of hPSC-BAOs to different concentrations of CSE (1%, 2%, and 4%). Even exposure to 1% CSE produced NO, which progressively increased in a manner dependent on the dose of CSE (Fig. 4A). We also found that 1% CSE exposure induced oxidative stress and inflammatory responses. The gene expression levels of tumor necrosis factor-alpha (TNF-α), IL-1α, IL-8, and IL-10 significantly increased in response to CSE exposure. Genes related to oxidative stress (NADPH oxidase 1-4 [NOX1-4]) and endoplasmic reticulum (ER) stress were also significantly upregulated (Fig. 4B and 4C). These results indicate that CSE induces an inflammatory response through the generation of pro-inflammatory mediators in hPSC-BAOs, suggesting that hPSC-BAOs could be an alternative to the CSE-mediated COPD model.
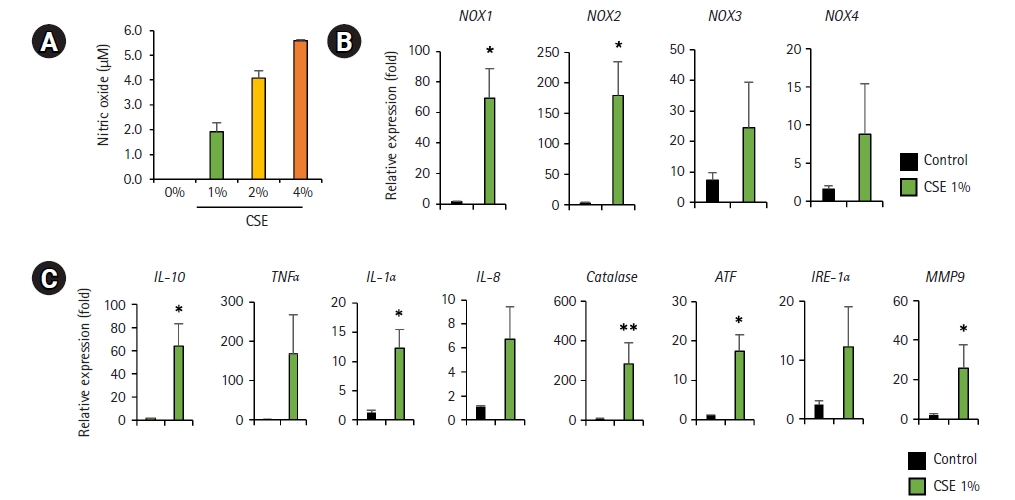
Nitric oxide (NO) production, NOX expression, endoplasmic reticulum (ER) stress, and inflammatory response to cigarette smoke extract (CSE) exposure in human pluripotent stem cell (hPSC)-bronchoalveolar organoids (BAOs). (A) NO production following CSE exposure in hPSC- BAOs was measured by NO assay. (B) NOX gene expression in 1% CSE-treated hPSC-BAOs. Data are presented as mean±standard deviation. (C) Expression of inflammation and ER stress-related genes in 1% CSE-treated hPSC-BAOs. Data are presented as mean±standard deviation. *p<0.05, **p<0.01.
Discussion
In recent years, 3D BAO system-derived murine and human lung tissues have been developed as a physiologically relevant platform for in vitro modeling of early lung development and pathogenesis. Rowbotham et al. demonstrated that chromatin alterations in aged mouse lungs influenced the frequency and activity of lung progenitor populations, leading to low efficiency of BAO formation [20]. Using a murine BAO system, Leeman et al. [21] showed that mesenchymal stem cells played an important role in protecting against hyperoxic lung injury by increasing the number of EpCAM+ stem cell antigen-1 (Sca-1)+ subsets. Furthermore, the activation of Wnt/β-catenin signaling in mouse distal lung epithelial progenitors promotes the capacity of BAO formation [22]. More recently, a more complex and sophisticated BAO system has been developed by the engraftment of tissue-resident alveolar macrophages, which are essential components of the alveolar niche that establish a bidirectional communication with epithelial cells in BAO structures [23]. In addition, human fetal lung-derived BAOs have been successfully demonstrated as a model for severe acute respiratory syndrome coronavirus 2 infection [24].
In the present study, we generated hPSC-derived BAOs, which were composed of diverse cell types of alveolar and bronchiole tissues, and assessed their potential for modeling PF and COPD by exposure to TGF-β and CSE. TGF-β acts as a master switch for the induction of fibrosis and contributes to chronic inflammation and a marked increase in mesenchymal phenotype markers such as collagen, vimentin, and α-SMA [25,26]. We showed that TGF-β1 treatment of our hPSC-BAOs was sufficient to stimulate the epithelial-to-mesenchymal transition and inflammatory responses. Chronic inflammation and oxidative and ER stress play an important role in the development of COPD [10,27]. We found that CSE exposure to hPSC-BAOs increased the expression of NOX1 and NOX2, which are endogenous pro-oxidative enzymes. Additionally, inflammation- and ER stress-related genes were upregulated in CSE-treated BAOs compared to non-treated BAOs. Similarly, mouse lung tissue-derived airway and alveolar organoids have been treated with CSE, which impaired the ability of organoid formation and induced ER stress and apoptosis (34156033). These findings suggest that hPSC-BAOs could be a useful tool for recapitulating major features of PF and COPD and could be used for drug screening and gaining a better understanding of pathological development.
A suitable disease model requires not only disease induction, but also validation of the safety and effectiveness of currently used drugs (e.g., PF therapeutics such as pirfenidone and nintedanib) [17,18,28]. More importantly, PF and COPD interact not only with the respiratory system but with multiple other systems, including the immune and vascular systems. Modeling more complex endpoints and investigating the underlying mechanisms by which tobacco smoke and other substances cause disease will require more complex co-culture systems that model interactions between different cell types. We consider the co-culture of hPSC-BAOs with immune and vascular cells a research topic useful for establishing an optimized disease model.
Notes
Conflict of interest
No potential conflict of interest relevant to this article was reported.
Funding
This study was supported in part by the Experts Training Graduate Program for Particulate Matter Management from the Ministry of Environment, Korea and the Bio & Medical Technology Development Program of the National Research Foundation (NRF) funded by the Korean government (MSIT) (2017M3A9B3061838).
Author contributions
Conceptualization: SHH; Data curation: SJ, JHK, RR; Formal analysis: SJ, JHK, RR; Project administration: SHH; Investigation: SJ; Supervision: SHH; Writing-original draft: SJ; Writing-review&editing: SHH.
Data availability
Please contact the corresponding author for data availability.