Lung organoids: target cells for understanding respiratory diseases
Article information

Abstract
Human respiratory disease research currently lacks in vitro models that recapitulate most of the physiology and architecture of the lung. Furthermore, the complex composition and structure of the lung, as well as anatomical differences between humans and mice, frequently lead to disappointing results applied in vivo. Recent advances in organoid technology include new, sophisticated in vitro culture tools that have stimulated considerable interest due to their potential ability to functionally mimic the organ rather than two-dimensional culture or animal models. Hence, pluripotent stem cell-based organoid studies are emerging as an alternative approach able to recapitulate tissue architecture with remarkable fidelity. Moreover, these biomimetic tissue models can be used to investigate the mechanisms of progression of various diseases. Idiopathic pulmonary fibrosis (IPF) and chronic obstructive pulmonary disease (COPD) are the most severe multifactorial respiratory disorders, characterized by irreversible airflow and progressive disease in elderly people. These diseases exhibit a progressive loss of alveolar type 2 epithelial (AT2) cells and accumulation of macrophages in the alveoli, leading to impaired pulmonary function. Despite recent advances in the study of COPD and IPF, effective treatments are lacking because our understanding of those diseases is hindered by their unknown mechanisms. Thus, in this review, the role of AT2 cells and macrophages is highlighted, along with their cell sources and applications for IPF and COPD modeling.
Introduction
The lung is a highly complex and dynamic organ. In its proximal part, the bronchiolar airways are connected to a single tracheal tube. Moving distally, the bronchiolar airways gradually narrow and branch into bronchioles. The terminal bronchioles, as the most distal segments, are the last conducting airway and give rise to two or more respiratory bronchioles. Each respiratory bronchiole gives rise to three alveolar ducts, which are lined with alveoli, and the alveolar ducts terminate in alveolar sacs and alveoli (Fig. 1) [1]. The enormously complex lung tissue, with its hierarchical structure, includes approximately 40 different resident cell types needed to accomplish successful respiration [2]. The resident cells of the respiratory tract are classified into three main compartments: (1) airway cells in the bronchi, (2) alveolar unit cells, and (3) pulmonary vascular cells. Researchers interested in recapitulating lung organogenesis in culture are faced with the choice of how to mimic real biological cells. In light of the presence of roughly 40 different resident cell types, it is challenging to select the most functional cell types for the generation of organoids.
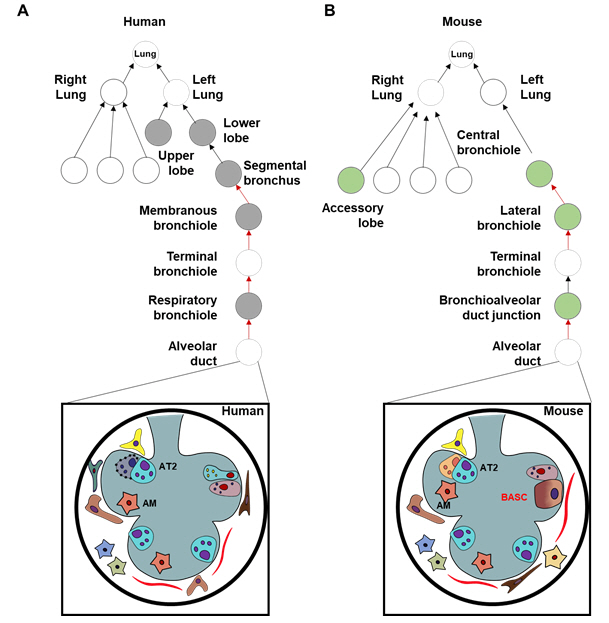
Comparative structure of the respiratory tract in humans and mice.
There are differences in the anatomical organization between the (A) human and (B) mouse lung. For example, differences are observed in the size, shape, and the number of lobes. The human lung has two lobes on the left and three lobes on the right, whereas the mouse lung has one lobe on the left and four on the right. The human lung has multiple, intrapulmonary, segmental bronchi with cartilage and submucosal glands. In mice, the cartilaginous airways end with lobar bronchi, and the lung is not subdivided into small units, unlike in humans. The cellular composition is also different between humans and mice. Briefly, the bronchoalveolar duct junction is located between the alveoli and distal bronchioles. Moreover, bronchoalveolar stem cells have the ability to differentiate into alveolar and bronchiolar lineages, bronchoalveolar stem cells is a specific alveolar and bronchiolar structures that are present in mice, but not humans. Basal cells are located in the distal region of the human lung, but are restricted to the trachea in the mouse lung. AT2, alveolar type 2 epithelial; AM,**; BASC,**.
With the development of protocols using human tissue stem cells and induced pluripotent stem cells (iPSCs) to study pathogenesis and ontogeny, lung organoid research is emerging as a valuable tool for respiratory disease modeling. Respiratory diseases, as well as lung cancer, are a major cause of morbidity and mortality in vulnerable human populations. Idiopathic pulmonary fibrosis (IPF) is a chronic disorder characterized by a progressive decline in pulmonary function and fibrosis [3]. IPF is a frequent and severe type of idiopathic interstitial pneumonia with an unknown etiology. It is characterized by progressive worsening of dyspnea and declining pulmonary function, and it has a median survival of approximately 2 to 3 years after diagnosis. Both the prevalence and incidence of IPF have consistently increased in recent years; hence, it is considered that severe exposure to particulate matter (PM), smoking, and viral or bacterial respiratory infections may be risk factors for IPF [4]. Since prednisone and azathioprine exhibited a lack of efficacy, pirfenidone with an anti-fibrotic agent has been approved for therapeutic use in patients with IPF. Generally, IPF has been recognized as an inflammatory-derived disease involving a back-and-forth process of lung injury and remodeling; however, an anti-inflammatory approach in isolation did not prolong the survival of patients with IPF. Currently, the pathophysiological perspective of IPF is slowly shifting to viewing the condition as an epithelial-driven and fibroblast-activated process leading to inflammation. Furthermore, chronic obstructive pulmonary disease (COPD) is the third leading cause of death worldwide. COPD is characterized by the progressive loss of functional parenchymal lung tissues, and thus the loss of alveolar gas exchange. In developing countries, the incidence of COPD is expected to increase over time in parallel with the prevalence of smoking in women and men. COPD involves sustained inflammation of the airways and lung parenchyma, and shows acute exacerbations. Since COPD and IPF represent major causes of disease and death, there is a desperate need for new effective tools. Thus, in this context, the role of alveolar type II epithelial (AT2) cells and macrophages as major cellular compartments will be discussed.
Ethics statement: This study was a literature review of previously published studies and was therefore exempt from institutional review board approval.
Disease mechanisms of IPF
1. Dysfunction of alveolar epithelium
The pathogenic mechanisms of IPF are uncertain; however, a currently favored concept is that in a genetically susceptible individual, recurrent environmental factors cause injuries, resulting in increased cell death, aberrant epithelial repair, and dysregulated epithelial and mesenchymal cellular networking, and promoting the activation of residential mesenchymal cells and extracellular matrix (ECM) accumulation [5]. In the lung, the alveolar epithelium lining is maintained by alveolar type I (AT1) and AT2 cells, which are essential for alveolar gas exchange and form a tight barrier to the passive leakage of fluid from the interstitium and vascular compartments into the alveolar space [6]. AT2 cells serve as the progenitor cells for alveolar regeneration during tissue repair and homeostasis, and they differentiate into AT1 cells in pathologic states including DNA repair and senescence [7,8]. Recently, Kobayashi et al. [9] have reported that AT2 cells undergoing differentiation into AT1 cells acquire pre-AT1 transitional cell states via TP53, transforming growth factor (TGF)-β, and DNA damage response signaling after alveolar injury. In human lungs with IPF, a single-cell RNA sequencing analysis revealed high enrichment of SFN, CLDN4, and KRT17 in a distinct cell cluster in pre-AT1 transitional cell states. This finding suggests that pre-AT1 transitional cells in the AT2 and AT1 transition process experience and respond to DNA damage, which is associated with degenerative lung diseases. AT2 cells show high expression of SFTPC and SFTP, which are capable of modulating the biophysical activity of pulmonary surfactant. Recently, mutation or deficiency of SFTPC, SFTPB, and ABCA3 in AT2 cells was shown to cause neonatal respiratory stress or early-onset interstitial lung disease [10]. In familial interstitial pneumonia, the expression of SFTPC with the L188Q mutation was found to alter the phenotype of AT2 cells through upregulation of endoplasmic reticulum (ER) stress and the unfolded protein response (UPR), suggesting that UPR activation contributes to fibrotic remodeling in IPF [11]. In IER-SP-CI73T mice, mutation of SPC triggered a rapid increase in mortality, and induced polycellular alveolitis leading to spontaneous fibrotic lung remodeling [12]. Moreover, SP-CI73T expression induced focal AT2 cell hyperplasia, exhibiting a persistent 40% increase in HA+/Ki-67+ cells, which may be an upstream driver of early lung injury. These findings support that certain components of the inflammatory response in the epithelium necessarily appear prior to fibrogenesis, and that SFTPC mutants recapitulate pivotal features of the human IPF phenotype. Indeed, patients with IPF frequently exhibit the atypical metaplastic epithelia in the fibrotic lung tissue, and showed increased p53 protein levels and DNA ploidy in the honeycomb region of IPF [13]. Recently, cigarette smoke-mediated AT2 cellular senescence was shown to reduce autophagy and to cause DNA damage-induced PARP1 activation, leading to pulmonary fibrosis [14]. In a comparison between control donor proximal and distal lung tissues and IPF fibrotic lung tissues, CDKN1A/p21, CDKN2A/p16, TP53, MDM2, and CCDN1 were significantly increased in AT2 cell transcriptomes, and the conditional loss of Sin3a was related to silencing of p53 expression, promoting AT2 cell dysfunction and driving progressive fibrosis [15]. Therefore, AT2 cell senescence plays a key role in declining pulmonary function and the pathological features of IPF.
2. Aberrant macrophages
The lung is a representative organ responsible for pathogen clearance and the reinstatement of tissue homeostasis [16,17]. Macrophages are major cellular components that sense pathogens and mediate phagocytosis during all phases of pulmonary fibrosis. Macrophage dysfunction leads to a failure to clear apoptotic cells, resulting in an accumulation of autoantigen and increased sustained inflammation [18]. When pulmonary macrophages were depleted by liposomal clodronate in bleomycin-induced mice, macrophages had no effect during the inflammatory phase [19]. However, depletion of macrophages reduced the surrogate markers of fibrosis α-smooth muscle actin (α-SMA) and Col1A1, indicating that macrophages play a resolution-promoting role during the reversible phase of fibrosis. Gao et al. [20] have shown that netrin-1 derived from macrophages, but not fibroblasts, induces collagen accumulation in ColIα2-CreER mice, and therapeutic α1-adrenoreceptor antagonism suppressed fibrosis. In addition, IPF macrophages showed enriched netrin-1 expression and 22 patients using α1-blockers exhibited significantly better lung function in terms of forced vital capacity and diffusing capacity for carbon monoxide. In Fra-2Tg mice, alternatively activated macrophages were dominant in cells of the macrophage linage expressing colony-stimulating factor 1 receptor and SPC-positive AT2 cells. They exhibited prefibrotic genes including periostin, α-SMA, and fibroblast-specific protein 1 (S100a4); however, Fra-2△Ad mice showed limited Fra-2 expression in AT2 cells and milder lung fibrosis progression [21].
In recent studies, nintedanib and pirfenidone have been pursued to treat mild to moderate IPF. However, the major treatments are limited due to side effects; moreover, the mechanisms of these drugs mainly focus on anti-fibrotic effects. IPF is a progressive and sustained fibrotic lung disease that primarily affects older adults [22]. Aged lungs undergo a degenerative process through the accumulation of extrinsic and intrinsic damage, which results in altered cellular homeostasis and apoptosis. Moreover, aging causes the decline of mitochondrial function and the mitochondrial respiratory chain, while increasing reactive oxidative stress (ROS) production, which is associated with mitochondrial DNA damage [23]. Damaged mitochondria with PINK1 deficiency and insufficient mitophagy accelerated the fibrotic progression of AT2 cells [24]. In alveolar macrophages, mitochondrial calcium uniporter, which triggers a Ca2+ influx, caused profibrotic polarization and augmented mitochondrial ROS production [25]. In patients with IPF, oxidative phosphorylation is decreased in primary alveolar macrophages, while the production of mitochondrial ROS is increased [26]. Although LC3B (an autophagy marker) and the selective autophagy receptor p62 were not changed in IPF alveolar macrophages, an imbalance in mitochondria biogenesis and degradation is involved in the pathogenesis of IPF.
Recent studies have found that age-related changes are associated with the differential severity of pneumonia in older patients. In the alveolar macrophage transcriptome, advanced age was associated with a reduced expression of cell-cycle related genes and the aged microenvironment of macrophages conferred resistance to the granulocyte macrophage colony-stimulating factor axis [27]. When tissue-resident alveolar macrophages from old and young adult mice were analyzed, aging-associated changes in the transcriptome of macrophages were entirely attributable to the alveolar environment, indicating that the cellular interactions of macrophages and AT2 cells are essential to restore youthful responses in the aged lung.
Disease mechanism of COPD
COPD is an irreversible and progressive disease of the lung following a pulmonary inflammatory response, leading to shortness of breath, chronic cough, and phlegm production [28,29]. Pulmonary inflammation is mediated by several stimuli, such as oxidative stress, aging, and defense against viral/bacterial infections [30]. COPD consists of two distinct pathological patterns: chronic obstructive bronchitis and emphysema. In the proximal region, inflammation and excessive mucus infiltrate, resulting in a narrowing airway with alveolar wall thickening [31]. In the distal region, alveolar walls composed of alveolar epithelial cells, including AT1 and AT2, are destroyed, causing failure of gas exchange (emphysema) [7]. Biological, cellular, and molecular characteristics all play a considerable role in the pathogenesis of COPD.
1. Oxidative stress and inflammation
Cigarette smoking, which is known as a major cause of COPD, results in a high level of ROS, which activate airway/alveolar inflammation and contribute to disease development [32]. Prolonged high levels of ROS can exacerbate COPD pathogenesis. Indeed, ROS signaling is intimately associated with cell damage, including cell death, apoptosis, aging, and mutations [33]. Cigarette smoke-induced oxidative stress regulates redox-sensitive transcription factors, such as mitogen-activated protein kinase (MAPK) family, nuclear factor kappa-B (NF-κB), and signal transducer and activator of transcription 1 (STAT1) [34]. These factors stimulate them, leading to the further recruitment of inflammatory cells in the alveoli. In COPD patients, excessive numbers of inflammatory cells, especially macrophages, are observed in the bronchoalveolar lavage fluid (BALF) and airways. They produce pro-inflammatory cytokines such as interleukin (IL)-1β, IL-6, tumor necrosis factor-alpha (TNF-α), and monocyte chemoattractant protein-1.
2. Matrix remodeling
In addition, IL-1β increases the expression of neutrophilic cytokines and matrix metalloproteinases (MMPs), which are proteolytic enzymes that degrade the matrix components [35,36]. The pathogenesis of COPD is related to imbalanced protease/antiprotease levels, which are essential for maintaining the elastin framework [34]. It has been suggested that the destruction of connective tissue by various proteases contributes to emphysema. In normal tissue, MMPs are tightly regulated by tissue inhibitor of metallopeptidase, which maintains a well-balanced state [37]. It has been suggested that disruption of the balance between apoptosis and replenishment of structural cells in alveoli contributes to the destruction of lung tissue in response to injury [38].
3. Alveolar epithelial apoptosis
As discussed above, pulmonary emphysema is characterized by epithelial cell death. In emphysema, destruction of epithelial cells caused by apoptosis leads to airspace enlargement, in which gas exchange fails, resulting in dyspnea [39]. Alveolar epithelial apoptosis is mediated by several mechanisms, including disruption of the balance between proteolytic and anti-proteolytic molecules, inflammation, oxidative stress, inhibition of vascular endothelial growth factor (VEGF) receptor, regulation of autophagy, and cell cycle arrest [38,40-42]. A VEGF-receptor 2 inhibitor, SU5416, led to a progressive increase in caspase-3 activity in rat lungs [40]. Caspase-3, a cascade protein, is considered to be a hallmark of apoptosis [43]. Furthermore, autophagy, a homeostatic process of intracellular degradation system, has been reported to be deeply involved in apoptosis signaling [44]. According to other research, autophagic cell death is caused by ROS resulting from autophagic degradation by catalase [45]. In general, autophagy is considered to play a protective role in normal tissue; however, prolonged and excessive autophagy has also been associated with autophagic cell death [46]. It has been reported that autophagy plays a functional role in stress responses to cigarette smoke exposure in vitro [42]. Both in vitro and in vivo, cigarette smoke-induced autophagy in alveolar epithelial cells. Qin et al. [47] showed that nuclear receptor 77 mediated CS-induced autophagy, which increased apoptosis in A549 cells and mouse.
Wei et al. [48] presented the gene-expression profile in COPD. They performed a microarray analysis using COPD patient samples to investigate the gene-expression patterns of COPD and to explore possible therapeutic targets. As described in the paper, the upregulated genes were involved in the immune response, antigen processing and presentation, positive regulation of immune response, and negative regulation of caspase activity, while the downregulated genes were significantly related to the functions of response to drugs, regulation of transcription, and especially steroid metabolic processes. In particular, HLA-A expression was significantly increased in COPD; therefore, they suggested that HLA-A may serve as a diagnostic marker for COPD. In our previous study, we found that the receptor for advanced glycan end products (RAGE) pathway was related with COPD pathology [49,50]. Then, we showed that a RAGE antagonist, FPS-ZM1, can be therapeutic agent via blockade of RAGE–damage-associated molecular patterns (DAMP) signaling.
In animal disease models, several studies have used porcine pancreatic elastase (PPE) as a COPD inducer. PPE is a protease that can cause a protease-antiprotease imbalance and has been applied to induce emphysema and COPD in animals [51]. Although PPE successfully induced COPD-like pathology in mice, as shown by the inflammatory response, loss of epithelial cells, ROS levels, and COPD-related gene expression, it has limitations for mimicking human COPD. Recently, β2-agonists, long-acting muscarinic antagonists, and inhaled corticosteroids have been developed as drugs for COPD [52]. The above-mentioned drugs are mostly bronchodilators; thus, although attenuate symptoms of COPD such as phlegm, cough, and dyspnea, disease progression is not affected. Additionally, a selective phosphodiesterase 4 inhibitor, roflumilast, was also approached as COPD drug with an anti-inflammatory effect [53]. Although many studies of COPD cures have been tried, no therapy has successfully repaired the damaged alveoli or airway. Three-dimensional (3D) organoid systems are expected to be used in the fields of disease modeling, drug screening, and pre-clinical research for human COPD in vitro.
AT2 cells in respiratory diseases
To characterize the resident cells in the respiratory tract, cell-type specific markers have been identified that provide molecular definitions, which will eventually lead to optimal technology to distinguish genomic factors through transcription profiling by single-cell RNA sequencing [54]. Travaglini et al. [54] acquired normal lung tissue from bronchi (proximal), bronchioles (medial), and alveoli (distal), and grouped cells based on the expression of compartment-specific markers. In the three canonical lung cell types of the epithelial, endothelial and stromal compartments, they suggested that AT2 cells play a differential role in producing surfactant, which prevents alveolar collapse. In the lung, AT2 cells are specialized to produce surfactant, and considered the progenitor cells for the alveolar epithelium. Indeed, despite accounting for approximately 5% of the surface area, they play a diverse role including self-renewal, removing injured AT2 cells, proliferation, and cell-differentiation into AT1 cells for alveolar remodeling. Given the functional importance of AT2 cells, they should be used for in vitro 3D lung modeling, especially of the distal region.
1. Idiopathic pulmonary fibrosis
Recent studies have shown that dysregulated AT2 cells are highly involved in fibrotic progression [55]. It has been demonstrated that the stimulated AT2 cells have an impaired function of differentiation into healthy and mature AT1 cells, and that alveolar remodeling is initiated via a sub-fibrotic cell state from AT2 cells as a repair process leading to fibrosis [9,56]. Indeed, direct conversion into myofibroblasts, known as the epithelial-mesenchymal transition (EMT), is also considered a key IPF characteristic. During EMT, AT2 cells lose their typical cell functions, including differentiation, proliferation, migration, and cell-cell junction [57]. Previously, we reported that a mediator of IPF in vivo, bleomycin, induced apoptosis in AT2 cells with increased levels of Schlafen 1 (SLFN1), SLFN2, and SLFN4, which are involved in cell cycle arrest and apoptosis [58]. Wu et al. [55] demonstrated that impaired regeneration of AT2 after damage resulted in sustained mechanical tension. This pathological stimulation activated TGF-β signaling, a well-known factor related to the EMT, leading to an accumulation of myofibroblasts. At an early state of IPF progression, inflammation has been observed in animal models. Recent studies have shown that many inflammatory cells, such as macrophages, neutrophils and lymphocytes, contribute to the pathogenesis of IPF [59]. As inflammatory responses are mediated by various routes, AT2 cells repair damaged tissue via remodeling, which contributes to physiological changes, including accumulation of ECM, contraction of fibroblasts, tissue tension, and stiffness [60,61]. Sun et al. [61] showed that tankyrase inhibition could impair AT2-AT1 differentiation via TAZ inhibition using an AT2 organoid system. They found decreased RAGE expression, furnishing evidence of a chronically inadequate capacity to regenerate AT2 in IPF in a system that could even be representative of human patients. Although AT2 cells play a critical role in lung repair, direct molecular targets remain to be elucidated.
2. Chronic obstructive pulmonary disease
In distal region of lung tissue, the alveolar epithelium (where gas exchange typically occurs) protects against the external environment, including PM, viral or bacterial infections, and even harmful factors from cigarette smoke. Chronic respiratory injuries result in a loss of epithelial cells, leading to shortness of breath. This pathological condition is known as emphysema, which is a component of COPD [62]. Cigarette smoking, a major risk factor for COPD, causes high levels of free radicals, which increase oxidative stress in AT2 cells [50]. ROS are believed to be a mediator in the development of COPD, through their involvement in apoptosis, aging, mitochondrial damage, and inflammation in AT2 cells [63].
Previously, we established a two-dimensional (2D) in vitro COPD model involving cigarette smoke extract (CSE). CSE exposure induced inflammation and ROS production. Interestingly, we identified high expression of RAGE via DAMP-Nrf signaling in COPD-like damaged AT2 cells. In both in vivo and in vitro studies, a RAGE antagonist (FPS-ZM1) showed an efficient curative effect via blocking RAGE-mediated Nrf2/DAMP signaling in damaged AT2 cells [49,50]. Likewise, it is important to find a novel disease-associated target that might allow a curative approach in vitro. Since 3D organoids can recapitulate the cellular diversity of complex lung tissues, unlike 2D systems, many researchers have expected that 3D lung organoid systems will serve as a useful modeling tool for pathologic or therapeutic studies in the field of pulmonary diseases. Nevertheless, it is necessary to develop models with a greater similarity to the human lung environment to recapitulate disease conditions or homeostasis against damage in vitro.
Macrophages in respiratory diseases
Macrophages are a critical cellular component of the innate immune system, and they are the first line of defense against pathogens in several tissues, including the brain (microglia), liver (Kupffer cells), intestine (lamina propria macrophages) and lungs (alveolar macrophages) [64]. In the lung, macrophages are the most abundant type of immune cell, and their heterogeneity involves the maintenance of pulmonary homeostasis, removal of cellular debris, microbial clearance, and resolution of inflammation [65]. Lung macrophages in the alveoli were previously thought to be a homogeneous population, but research in the last decade has shown that they are highly heterogeneous in their origins, functions, and phenotypes. There are two major subsets. One is the well-described category of alveolar macrophages, which populate the alveolar and airway lumen. The other subset, interstitial macrophages, is less well known due to their diffuse localization within the lung parenchyma.
Alveolar macrophages, the resident mononuclear phagocytes of the lung, are situated at the air-tissue interface. It has been clearly demonstrated that the alveolar macrophages play a critical role in the maintenance of homeostasis in defense against microbes (such as viruses, bacteria, and fungi), inhaled environmental particles (e.g., coal, silica, and asbestos), tissue debris, and cancer cells. Macrophages are highly plastic cells able to display versatile functional phenotypes in response to micro-environmental stimuli. Their plastic characteristics refer to macrophage polarization, a process in which macrophages differentiate into specific phenotypes that exhibit certain biological functions. There are two major phenotypes of alveolar macrophages: the classically activated (M1) macrophages and the alternatively activated (M2) macrophages. Unfortunately, the two subsets are indistinguishable by surface morphology; however, they exhibit significant transcriptional, epigenetic, and functional differences.
M1 macrophages respond to microbial factors or Th1 pro-inflammatory cytokines, exhibiting a glycolytic metabolism and enhanced phagocytic and bactericidal activity. Lipopolysaccharide and Th1 cytokines, including interferon-delta and TNF-α, cause polarization into M1 macrophages. M1 macrophages physiologically participate in the removal of pathogens, and ROS and nicotinamide adenine dinucleotide phosphate are subsequently upregulated. In contrast, M2 macrophage polarization is caused by Th2 cytokines; these macrophages undergo oxidative metabolism via anti-inflammatory cytokines, perform phagocytosis, and deposit collagen during recovery after injury. M2 polarization occurs through Th2 cytokines such as IL-4, IL-13, IL-10, IL-33, and TGF-β [66]. M2-polarized macrophages express high levels of CD206, decoy receptor IL-1 receptor II, and IL-1 receptor antagonist. With their remarkable plasticity, alveolar macrophages are highly sensitive in response to micro-environmental signaling, and functionally participate in the pathogenesis of respiratory diseases.
Interstitial macrophages are relatively unknown compared to alveolar macrophages due to their histological localization. In the steady-state, lung interstitial macrophages are primarily considered as “non-alveolar” macrophages and are located in the bronchial interstitium and interstitial space of the alveolar septum or alveolar corners. Alveolar macrophages rapidly patrol the alveolar space, sensing pathogenic stimuli, whereas interstitial macrophages are proportionally less abundant and exhibit lower phagocytic potential in response to inflammatory stimuli [67]. Recently, Gibbings et al. [68] demonstrated that bronchial interstitial macrophages (BIMs) are classified into three subsets: BIM1 (CD11cloMHCIIlo), BIM2 (CD11cloMHCIIhi), and BIM3 (CD11cloMHCIIlo). The BIMs have phagocytic potential, and show the expression of monocyte-specific markers such as CD14, CD163 and Csfr1. Interstitial macrophages are emerging as a pivotal modulator in the maintenance of homeostasis in the alveolar and bronchial regions.
1. Idiopathic pulmonary fibrosis
As discussed above, IPF occurs when repetitive alveolar injuries induce the activation of mesenchymal cells and fibroblasts and the differentiation of alveolar epithelial cells into myofibroblasts. Repeated alveolar epithelial injuries cause an aberrant wound healing process leading to fibrotic plaque formation and excessive accumulation of ECM. In this process, macrophages acquire a phenotype that promotes fibro-proliferation in injured site. In particular, alveolar macrophages actively participate in ECM remodeling via increased MMPs of neutrophils and accumulation of collagen. Moreover, the activated alveolar macrophages release ROS and TGF-β. In the pathogenesis of IPF, sustained inflammation triggers the initiation of fibrotic responses with increased numbers of Ly6Chi monocytes. In macrophages of interferon regulatory factor 5 (IRF-5) deficient mice, the inhibition of IRF-5 associated with TGF-β led to fibrotic responses in adipose tissue, indicating the dual role of M1 macrophages in wound repair and fibrotic responses [69]. In general, infiltration of M2 macrophages is recognized as a critical regulator of fibrogenesis. In mice treated with bleomycin, loss of Chop significantly attenuated TGF-β production, decreased the number of M2 macrophages, and reduced ER stress [70]. Recently, Cui et al. [71] reported that apolipoprotein E, produced primarily by monocyte-derived alveolar macrophages, promoted type I collagen phagocytosis and interacted with low-density lipoprotein receptor-related protein 1 leading to the resolution of lung fibrosis in IPF mice. They demonstrated that monocyte-derived alveolar macrophages have distinct functions in the early fibrogenic and fibrotic resolution phases. In contrast, interstitial macrophages are rapidly positioned in the interstitium to influence lung fibrotic processes, rather than alveolar macrophages. In bleomycin- and radiation-induced mice, CD206-expressing interstitial macrophages acquired a profibrotic phenotype and mediated the fibrogenic process, accelerating the myofibroblast differentiation. Interstitial macrophages isolated from tissues with radiation-induced lung fibrosis showed highly increased levels of arginase-1, and inhibition of colony-stimulating factor receptor-1 was significantly associated with a decrease of radiation-induced lung fibrosis [72]. Thus, alveolar and interstitial macrophages in the lungs show contrasting and distinctive phenotypes, and they play separate roles depending on the phase of IPF.
2. Chronic obstructive pulmonary disease
It is widely accepted that alveolar macrophages play a critical role in the pathogenesis of COPD. In patients with COPD, markedly increased numbers of alveolar macrophages were found in airways, lung parenchyma, BALF, and septum. The patients with COPD have high number of macrophages. In COPD mice, alveolar macrophages released elastolytic enzymes, proteases, MMP-2, MMP-9, and cathepsin K, leading to the destruction of alveoli [73]. Impairment of alveolar macrophages leads to an increased susceptibility to bacterial colonization and exacerbation by respiratory viruses or bacteria. M1/M2 macrophage polarization has been reported to determine the impact of COPD progression. A high ratio of nitric oxide–expressing M1 macrophages was observed in COPD airways, while arginase-expressing M2 macrophages were present in COPD mice. Hence, a better understanding is needed of the M1/M2 macrophage polarization state and the roles of macrophages in the pathogenesis of COPD. In COPD, the functions of interstitial macrophages are veiled compared with alveolar macrophages.
Modeling of IPF and COPD using 3D organoids
The coronavirus disease 2019 (COVID-19) pandemic has caused a spectrum of respiratory illnesses, extending to acute respiratory distress syndrome. Animal models, including mice expressing human ACE2, STAT-2 knockout hamsters, and ferrets with severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) infection, have been applied to study COVID-19; however, most of the animal models failed to recapitulate severe lung dysfunction [74-76]. The apparent limitations of animal models desperately need to be supplemented by applications of human alveolar cell compartments. Lamers et al. [77] showed that human lung bud tip organoids from the canalicular stage were grown and differentiated into alveolar-like cells in a bronchioalveolar-like air-liquid culture. When those organoids were infected with SARS-CoV-2, the differentiated cells expressed ACE2 and TMPRSS2. In addition, the differentiation of AT2 cells into AT1 cells during alveolar regeneration after fibrotic injury has been traced, and interstitial macrophage–derived IL-1β triggered reprogramming of AT2 cells in a 3D culture [56]. Recently, TGF-β caused fibrotic changes, including the ECM (COL1A1 and COL1A2), mesenchymal (vimentin), and EMT (α-SMA, CTNNB1, TWIST1, and SNAIL1) in iPSC-derived alveolar organoids, suggesting that AT2 cell-mediated organoids have a meaningful ability to respond to fibrotic stimuli, suggesting their ability to serve as platforms [78].
Conclusion
Although researchers hesitate to apply lung organoids due to the lung’s complex structure with a capillary network, lung organoids hold great potential in clinical translational research. Lung organoids have advantages in reproducing human tissue for modeling respiratory diseases in vitro. They are useful tools to determine pathological targets, making it easier to study possible therapeutic approaches. Although AT2 cells and macrophages were mainly discussed in this context, the identification of surface markers/receptors in other human lung compartments remains necessary (Fig. 2). Furthermore, the blending of lung organoids with 3D bio-printing and vasculature may facilitate models that recapitulate the alveolar microenvironment with the desired cellular composition for future disease modeling.
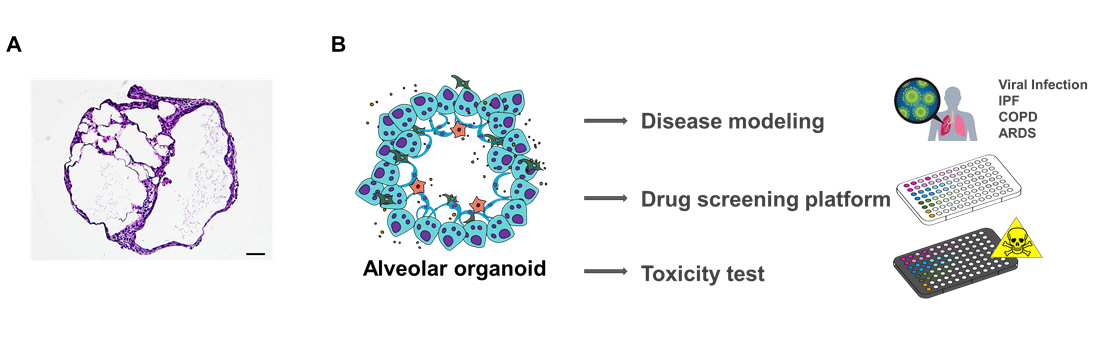
Applications of lung organoids.
An induced pluripotent stem cells–derived alveolar organoid is established for 25 days in vitro. (A) A histological image is analyzed under a microscope with hematoxylin and eosin staining. Scale bar: 50 μm. (B) Organoid systems are expected to serve as useful tools for modeling human diseases, including viral infections, chronic obstructive pulmonary disease (COPD), idiopathic pulmonary fibrosis (IPF), and acute respiratory distress syndrome (ARDS). They can be applied in the fields of drug screening and toxicity testing.
Notes
Conflict of interest
No potential conflict of interest relevant to this article was reported.
Funding
This study was supported by grants from the National Research Foundation (NRF) funded by the Korean government (MSIT, NRF-2020R1A2C2010712, 2020R1A6A3A13072437) and 2020-01-02-087 the National Institute of Environment Research (NIER).
Additional contributions
We thank Leeseol Choi and Sebi Lee (Department of Thoracic and Cardiovascular Surgery, Kangwon National University School of Medicine, Chuncheon, Korea) for technical assistance.
Data availability
Please contact the corresponding author for data availability.