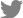Introduction
Type 1 diabetes is characterized by the autoimmune destruction of pancreatic β cells, necessitating lifelong insulin administration for disease management [1,2]. In contrast, type 2 diabetes occurs when β cells fail to meet increased insulin demands, which can lead to insulin administration in certain cases. In the late stages of type 2 diabetes, β -cell failure can also occur, requiring constant insulin administration [1,3]. While direct insulin injections can be life-saving, achieving tight glucose control remains difficult, often resulting in complications such as life-threatening hypoglycemia, as well as microvascular and macrovascular complications induced by hyperglycemia [1,4,5].
New therapeutic modalities, such as cell and gene therapies, have emerged as complementary approaches to conventional drugs for treating refractory diseases. Cell therapies, in particular, offer substantial potential for addressing type 1 and late-stage type 2 diabetes, as they allow the replacement of lost β cells through the transplantation of therapeutic cells into patients [6,7]. Given the ongoing increase in the number of diabetes patients worldwide [8], there is an urgent need to develop cell-based therapies to treat and potentially cure this refractory disease.
The original Edmonton protocol developed in the early 2000s and a recently approved cell therapy, Lantidra, both utilize pancreatic islets from organ donors to produce cell-based therapeutics [9,10]. However, the scarcity of organ donors limits the widespread adoption of this treatment approach. To address this challenge, insulin-producing β cells have been generated in vitro, primarily from pluripotent stem cells (PSCs) [6,7]. This process involves mimicking in vivo developmental pathways in cell cultures to guide PSCs toward the pancreatic lineage, ultimately yielding stem cell-derived β cells (SC-β cells) [6,7]. Currently, SC-β cells are the subject of extensive research as next-generation therapeutics for diabetes [11-13].
Despite the potential of SC-β cells, several critical challenges must be addressed to generate highly functional SC-β cells suitable for therapeutic applications. Notably, current SC-β cells exhibit functional immaturity, characterized by insufficient glucose-stimulated insulin secretion (GSIS) when compared to primary islets, although they can synthesize and store insulin in their secretory granules [14,15]. Furthermore, the transplantation of these cells may trigger immune responses in the recipient, leading to the complete elimination of the cell therapeutics. The risk of immune rejection necessitates the continuous administration of immunosuppressive drugs, which can give rise to various adverse effects [6,16]. In addition, transplanted cells could suffer from hypoxia and subsequent apoptosis due to inadequate vascularization [17]. Research has shown that about half of transplanted β cells may be lost within the first 7 days post-transplantation [18]. Given that β cells are particularly vulnerable to hypoxia due to their exceptionally high oxygen requirements [19], methods for protecting the transplanted β-cell mass from hypoxia are crucial to ensure maximal therapeutic effects. Furthermore, the transplantation of live cells raises safety concerns as they may develop into tumors or trigger severe immune responses [20-22]. To guarantee the safe administration of these novel therapeutic modalities, it is of the utmost importance to develop methods for precise control over the cells.
These challenges can be addressed through a variety of approaches, including the refinement of differentiation protocols, biomaterial engineering, and genome editing. In particular, current genome-editing tools based on the clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 system hold significant potential because they can permanently alter the genomic sequence and, consequently, the phenotype of cell therapeutics [23].
SC-β cells not only serve as cell therapeutics, but also as a crucial source of human β cells for investigating β-cell biology and unraveling the mechanisms of diabetes. Currently, functionally mature human β-cell lines are difficult to obtain [24], and researchers often resort to using rodent β-cell lines as substitutes, despite the evident differences between human and rodent islets [25]. Moreover, the availability of primary human islets for basic research is severely limited. Consequently, SC-β cells are emerging as indispensable tools for exploring human β cells and advancing diabetes research. CRISPR/Cas9-based genome editing is employed as a key technology to introduce genetic variants present in diabetes into SC-β cells, enabling precise investigations into the genetic aspects of the disease.
Here, we provide an overview of recent progress in engineering SC-β cells through genome editing. First, we briefly discuss methods for generating SC-β cells and state-of-the-art genome editing tools. We then delve into how these genome editing tools are being employed to enhance the function of SC-β cells for the development of cell-based therapeutics for diabetes. Next, we explore genome editing-based modeling of diabetes-related genetic variations in SC-β cells. Furthermore, we discuss recent efforts to engineer primary islets through genome editing (Table 1). Since the application of genome editing in β-cell therapeutics is still in its infancy, we present remaining tasks and outline future directions for the application of this approach in implementing cell-based therapeutics for diabetes.
Generation of stem cell-derived β cells
Ethics statement: This study was a literature review of previously published studies and was therefore exempt from institutional review board approval.
Two seminal protocols reported in 2014 outlined the generation of insulin-producing β-like cells from embryonic stem cells (ESCs) and induced PSCs (iPSCs), collectively referred to as PSCs in this review [26,27]. Since then, many researchers have relied on these protocols to generate SC-β cells. These protocols induce the differentiation of PSCs into stem cell-derived tissues using various molecules that modulate intracellular signaling pathways, directing the PSCs through specific developmental stages (Fig. 1).
Some of these cell-directing molecules are proteins, such as activin A, keratinocyte growth factor, and betacellulin, which act on membrane receptors [26,27]. The majority of cell-directing components consist of small molecules that act on diverse intracellular targets and modulate corresponding signaling pathways. For instance, in the first stage of the differentiation, CHIR99021 inhibits glycogen synthase kinase 3, activating the Wnt pathway and promoting stem cell differentiation into the endoderm [26-28]. Retinoic acid, a fundamental signaling molecule in vertebrate endoderm development, is employed to direct differentiation into pancreatic progenitors [26,27,29]. SANT1, an inhibitor of the Sonic hedgehog pathway, enhances pancreatic specification [26,27,30], while LDN193189, an inhibitor of the bone morphogenetic pathway, facilitates conversion into pancreatic endocrine precursors [26,27,31]. Alk5i, an inhibitor of the transforming growth factor-β type I receptor kinase (ALK5), and the small thyroid hormone T3 (L-3,3ʹ,5-triiodothyronine) are employed in the late stage of differentiation to generate insulin-positive β-like cells (Fig. 1) [26,27].
These universal protocols demonstrated the feasibility of generating SC-β cells for future therapeutic applications. However, these protocols have limitations, such as low yields of insulin-producing cells and insufficient GSIS by the resulting SC-β cells. To address these challenges, researchers have developed numerous improved protocols based on the universal protocols. For example, new small molecules have been employed to generate more mature SC-β cells by modulating alternative intracellular signaling pathways [32,33].
Hogrebe et al. [32] conducted RNA sequencing analysis to identify factors affecting the differentiation of PSCs into endodermal lineage. They discovered that polymerized cytoskeleton inhibited neurogenin 3 (NEUROG3)-induced endocrine differentiation. NEUROG3 expression should be initiated at a specific differentiation step to guide the cells into becoming SC-β cells, while its early expression results in the production of polyhormonal cells. Building on this insight, they investigated small-molecule modulators of actin polymerization and found that treating cells with latrunculin A, an actin filament destabilizer, at stage 5 resulted in increased NEUROG3 expression and the subsequent generation of SC-β cells with enhanced GSIS. Notably, the improved protocol allowed β-cell production using a 2-dimensional culture platform, simplifying production methods and reducing costs for lab-scale processes, thereby enabling the widespread use of SC-β cells [32,34].
β-cell maturation is linked to reduced proliferation, and aurora kinase is known to induce the proliferation of primary β cells [35]. Therefore, Balboa et al. [33] treated cells at the final stage of differentiation with ZM447439, an aurora kinase inhibitor, to promote the maturation of SC-β cells. This treatment reduced cell proliferation without affecting the fraction of insulin-expressing cells. Importantly, the compound-treated cells displayed substantially improved GSIS, underscoring the importance of suppressing cell proliferation at the final stage [33].
In another approach, the differentiation process was modified to recapitulate endocrine cell clustering, resulting in islet-like clusters that exhibited physiological properties analogous to those of primary human β cells [15].
Another promising approach for improving the quality of SC-β cells is genome editing. Recent advances in genome-editing technologies have enabled the introduction of any genetic material into the genome. This capability allows cells to be readily modified to harbor functional genes or correct pathogenic variants for use in therapeutics. In the next section, we briefly discuss recent genome-editing technologies widely used for basic research and therapeutics development.
Genome-editing technologies
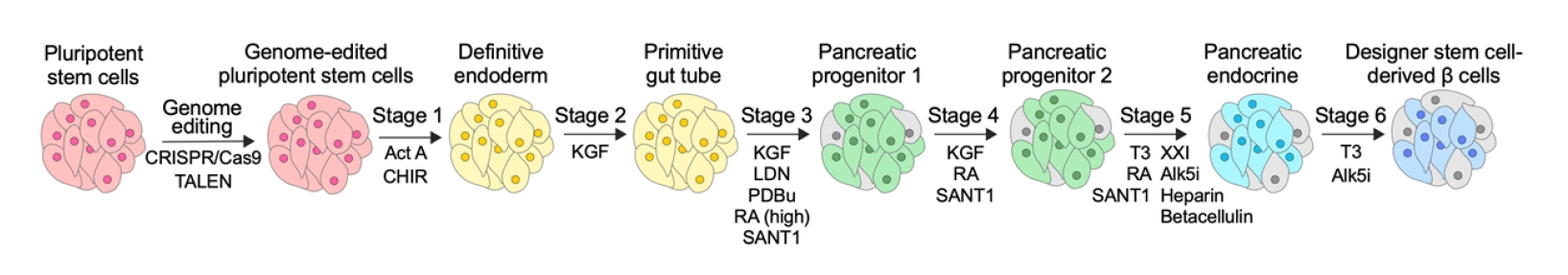
Rapid advancements in genome-editing technologies, particularly the CRISPR/Cas9 system, have enabled the facile modification of specific genes for elucidating biological phenomena and developing new therapeutic modalities. Cas9 generates a double-strand break (DSB) at the desired genomic locus, directed by specific guide RNAs (gRNAs). The resulting DSB is then repaired by cell’s own DNA repair pathways to join the broken ends (Fig. 2).
In the error-prone non-homologous end-joining (NHEJ) pathway, a small insertion or deletion is introduced into the DNA, ultimately leading to the knock-out of the target genes. When short homologous sequences are present on both sides of the DSB, the microhomology-mediated end-joining (MMEJ) pathway can lead to small deletions and consequent gene knock-out. For knock-in of specific sequences at the DSB, a donor DNA that contains homology arms and the desired sequence is delivered along with Cas9 and gRNA, which leads to homology-directed repair (HDR), facilitating the insertion of DNA sequences into the genome (Fig. 2). Typically, the knock-out efficiency by NHEJ and MMEJ is high, but the HDR efficiency is low. Consequently, a significant amount of research has been focused on enhancing HDR efficiency to enable precise editing in various cell types, including stem cells [36-38].
Different genomic loci can be targeted simply by changing the 20-nt gRNA sequence; therefore, CRISPR/Cas9 has been extensively applied for generating genetic modifications in various cells. This modular feature has allowed many researchers, including non-specialists, to use the CRISPR/Cas9 system effectively. Furthermore, CRISPR/Cas9 exhibits robustness across diverse biological systems. For instance, it can be utilized with various cell types, including cell lines and primary cells, and even across different organisms in vivo [23,39]. It can be delivered into cells through various forms, such as plasmids, mRNA, ribonucleoproteins, and viral vectors. This versatility allows researchers to choose the most suitable tools for their specific genome-editing scenarios.
The extensive application of CRISPR/Cas9 to genome editing in PSCs holds the promise of dramatic improvements in the performance of stem cell-derived therapies. Notably, the HDR pathway, which is essential for gene knock-in, is active only in dividing cells. This enables gene knock-in to be performed in PSCs, which can proliferate through cell division. However, SC-β cells, which rarely proliferate, do not allow gene knock-in via HDR [40,41]. As a result, a common approach involves initially editing PSCs to introduce genetic materials through the HDR pathway. The edited PSCs are then converted into SC-β cells (Fig. 1). In contrast, knock-out using the NHEJ pathway can be performed in both dividing and non-dividing cells. Therefore, target genes can be efficiently knocked out in PSCs, and the resulting cells can be converted into knock-out SC-β cells. In some cases, mature SC-β cells or even primary pancreatic islets can be directly edited for knock-out.
Genome editing to overcome challenges of current SC-β cells
In this section, we briefly discuss the major challenges encountered in the production of SC-β cells and introduce how genome-editing tools are being employed to overcome the issues.
A significant challenge with current protocols for generating insulin-producing β cells is that they often yield a mixture of diverse cell types, resulting in a low yield of pure insulin-producing β cells [42]. Therefore, it is crucial to purify the β cells from this heterogeneous cell mixture for therapeutic applications.
Lee et al. [43] employed genome editing to address this challenge by generating an iPSC line expressing the pancreas/duodenum homeobox protein 1 (PDX1)-enhanced green fluorescent protein (EGFP) fusion protein. PDX1 is a critical pancreatic β-cell marker essential for β-cell development and maturation [43]. Thus, functionally mature SC-β cells would express the PDX1-EGFP fusion protein. They used CRISPR/Cas9-initiated HDR to knock in the EGFP sequence at the C-terminus of PDX1 in an iPSC line. When these engineered iPSCs were differentiated into SC-β cells, EGFP expression was specifically detected in insulin-producing cells, but not in the definitive endoderm stage. This strategy holds promise for purifying mature β-cell therapeutics [43]. Yoshihara et al. [44] employed CRISPR/Cas9 to knock in the GFP sequence downstream of the INS promoter, enabling the facile observation of insulin-producing cells in iPSC-derived islet-like organoids.
Many recent reports on generating SC-β cells rely on engineered ESC lines where the INS locus was edited through homologous recombination without the aid of Cas9 [15,45]. In this case, the ESCs were engineered only in one allele of the INS locus by inserting the GFP sequence. This approach allows easy identification, purification, and subsequent conversion of insulin-producing cells into highly functional 3-dimensional β-cell clusters [15,45]. While the absolute level of insulin production may decrease because only one allele produces insulin while the other produces GFP, these edited ESCs have been used in a range of research endeavors for generating functional SC-β cells due to their ease of use [46-50].
Genome editing has also been used to address the issues of immaturity in current SC-β cells. For example, Ma et al. [49] identified ZnT8, a zinc transporter predominantly expressed in β cells, as a potential target for enhancing the functional maturity of SC-β cells, on the basis of previous studies conducted in rodent and human cells. To investigate the role of ZnT8 in SC-β cells, CRISPR/Cas9-based genome editing was performed in ESCs to knock out the SLC30A8 gene, which encodes ZnT8. Subsequently, these modified ESCs were differentiated into SC-β cells. This knock-out yielded promising results, as GSIS significantly improved. The enhancement is attributed to the alleviation of Zn-mediated inhibition of insulin secretion upon stimulation by glucose. Furthermore, the knock-out SC-β cells exhibited increased resistance to cell death triggered by glucotoxicity and lipotoxicity, achieved by a reduction in endoplasmic reticulum (ER) stress through zinc level modulation. Single-cell RNA-seq revealed that ZnT8 loss-of-function altered the gene expression profile toward a functionally mature state in SC-β cells. Moreover, the edited SC-β cells demonstrated their therapeutic potential by successfully restoring normoglycemia in streptozotocin-induced diabetic mice.
Building upon the observation that ZNF148, a zinc finger transcription factor, acts as a negative regulator of insulin secretion in rodent cells [51], de Klerk et al. [48] employed a knock-out strategy to target the ZNF148 gene in ESCs. Subsequently, these knock-out cells were differentiated into SC-β cells. Remarkably, the loss of ZNF148 function did not impair the process of β-cell specification during differentiation, as evidenced by the similar fraction of C-PEP+, NKX6.1+, PDX1+ cells in the knock-out cells and their wild-type counterparts. However, when these knock-out cells were subjected to high glucose challenges, they exhibited notably higher insulin secretion than the wild-type cells. This enhancement in insulin secretion was attributed to upregulation of annexin and S100 proteins, which are involved in the regulation of insulin trafficking and exocytosis.
In summary, these studies underscore the potential of simple knock-out strategies to functionally enhance β-cell therapeutics.
Another significant challenge in current cell-based therapeutics arises from graft rejection triggered by immune responses. To circumvent this issue, cells can be delivered to patients using encapsulation devices. Alginate is a commonly used material for microencapsulation devices, while artificial polymeric materials find application in macroencapsulation devices [52]. Although these encapsulation devices effectively shield the transplanted cells from host’s immune system, they can introduce a new challenge—namely, a foreign body reaction to the implanted materials, which hampers the practical application of encapsulation strategies. For example, recent clinical trials involving macroencapsulated SC-β cells have observed foreign body responses and subsequent fibrosis around the devices in most cases [11,13]. To solve these problems, researchers are turning to genome editing of PSCs to create hypoimmunogenic cells. This approach aims to minimize allograft rejection by the recipient’s immune system without the need for cell encapsulation or the administration of immunosuppressive drugs (Fig. 3A) [53].
As allograft rejection is primarily mediated by the recognition of human leukocyte antigens (HLAs) by T cells, one of the key strategies in cell engineering focuses on the ablation of these molecules. In this context, Hu et al. [54] conducted CRISPR/Cas9-mediated genome editing in iPSCs to knock out the B2M and CIITA genes for ablating HLA class I molecules and HLA class II molecules, respectively. As the depletion of B2M can lead to the “missing self” killing response by natural killer (NK) cells [54,55], they conducted screening to identify additional factors capable of inhibiting the NK-cell response. Among several transgenes tested, virally expressed CD47 was found to be highly effective as an immune checkpoint inhibitor for NK cells. The resulting hypoimmune iPSCs, characterized by B2M and CIITA knock-out, along with CD47 overexpression, were then differentiated into SC-β cells. Importantly, the hypoimmune editing did not compromise the differentiation capacity of the iPSCs. These hypoimmune SC-β cells exhibited remarkable survival and successfully restored normal glucose levels in immunocompetent allogeneic humanized mice. Notably, the team applied the same hypoimmune engineering to primary macaque islets and transplanted them into an allogeneic rhesus macaque model. In this context, the engineered islets demonstrated long-term survival, while the wild-type islets faced rejection within a week. This groundbreaking study highlights the potential of hypoimmune engineering as a viable approach for developing therapeutic SC-β cells [54].
Similarly, Gerace et al. [56] employed the B2M knock-out strategy in human ESCs (hESCs) to generate HLA-deficient SC-β cells. These engineered cells demonstrated resistance to allogenic immune destruction by peripheral blood mononuclear cells and NK cells in vitro. Although these cells were able to restore normoglycemia in a diabetic mouse model, they were eventually destroyed, albeit with a reduced rejection rate. As a complementary strategy, the researchers developed another genome editing approach to engineer cells to secrete 3 cytokines (interleukin [IL]-2 N88D mutein, transforming growth factor [TGF]-β, and IL-10) that promote localized immune tolerance by recruiting regulatory T cells (Tregs) to the grafts. The CRISPR/Cas9-based HDR pathway was employed in hESCs to knock in the genes for these cytokines at the C-terminal locus of GAPDH. Since GAPDH is a constitutively expressed gene, the knock-in at the GAPDH locus allowed persistent transgene expression. Indeed, the resulting engineered hESCs were differentiated into SC-β cells that constitutively secreted these cytokines. This engineering enabled the survival of the transplanted cells and the reversal of diabetes in vivo, with Tregs localized within the graft [56]. This study demonstrates the feasibility of local immune modulation for the long-term survival of allografts.
Another hypoimmune strategy was developed by Gravina et al. [57] to protect cell therapeutics from antibody-mediated rejection. Their approach involved the overexpression of CD64, a high-affinity receptor for IgG Fc, on cell membranes. This overexpression led to the capture of monomeric IgG Fc on CD64, rendering it inaccessible to effector cells or complement systems. When this CD64 overexpression strategy is combined with the previously mentioned hypoimmune engineering (B2M and CIITA knock-out, CD47 overexpression), the iPSC-derived endothelial cells became entirely immune-evasive to prevent anti-Rh(D)-mediated cytotoxicity. In contrast, cells without CD64 overexpression were susceptible to cell death. This CD64 overexpression has the potential to extend to SC-β cells that express HLA-A2 and have not undergone prior genome editing. Upon transduction with viral vectors to overexpress an intracellularly truncated analog of CD64 (CD64t), these cells demonstrated protection against killing by anti-HLA-A2 IgG1, while the unmodified SC-β cells faced rapid loss both in vitro and in vivo [57]. Looking ahead, we envision the possibility of obtaining completely immune-evasive SC-β cells through multiplex editing (B2M and CIITA knock-out, CD47 and CD64 overexpression) in the near future.
Hypoimmune engineering has found application in several other studies, each with slightly different strategies. For instance, Parent et al. [50] conducted multi-step genome editing in ESCs to knock-out all classical HLAs except HLA-A2, because HLA-A2 enables the retention of HLA-E, which is capable of inhibiting NK cell-mediated killing. Importantly, this hypoimmune engineering did not disrupt the differentiation potential of ESCs. The resulting SC-β cells were effectively protected from rejection mediated by both T cells and NK cells, as demonstrated in a humanized mouse model. In addition to hypoimmune editing, another genome editing technology was employed to introduce a gene of interest at the AAVS1 genomic safe harbor locus. The researchers utilized transcription activator-like effector nucleases (TALENs) as a genome editor to insert a luciferase gene at AAVS1, facilitating noninvasive monitoring of cell survival in vivo through bioluminescence imaging [50]. Since the AAVS1 locus is transcriptionally active and amenable to the expression of transgenes without compromising cellular fitness [58], the AAVS1 editing strategy offers a versatile means of introducing diverse functional genes into cell therapeutics. For example, Castro-Gutierrez et al. [47] generated immune-evasive ESCs by knocking out B2M with CRISPR/Cas9 and knocking in a doxycycline-inducible overexpression cassette (Tet-On system) for programmed death ligand 1 (PD-L1) at the AAVS1 locus using TALENs. Their engineered ESCs were efficiently differentiated into SC-β cells with a similar efficiency as unedited cells. Notably, the engineered SC-β cells displayed reduced stimulation of CD8 T cells in vitro upon doxycycline-induced PD-L1 expression [47]. Similarly, Leite et al. [59] employed B2M knock-out in iPSCs to decrease the expression of HLA class I molecules in differentiated SC-β cells. The resulting cells displayed reduced T-cell activation, confirming that T-cell activation is mediated by direct engagement of T-cell receptors with HLA-peptide complexes in SC-β cells [59].
These HLA knock-out strategies have the potential for widespread use in the development of cell-based therapeutics due to their effectiveness in reducing immune responses post-transplantation. Technically, knock-out cells can be readily isolated by flow cytometry, since HLA molecules are located on the plasma membrane. This feature allows many researchers to employ this hypoimmune engineering technique to create a wide range of cell-based therapeutics.
To identify mediators of immunogenicity against transplanted SC-β cells, Sintov et al. [60] conducted single-cell RNA sequencing and genome-wide CRISPR screening under inflammatory environments, revealing that genes in the interferon pathways were upregulated under inflammatory conditions. A detailed examination of the screen's findings highlighted that chemokine ligand 10 (CXCL10) was particularly implicated in immune-graft interactions. Therefore, CXCL10 was ablated by knocking in an EGFP gene into the middle of the CXCL10 locus in ESCs, and the edited ESCs were differentiated into SC-β cells. As anticipated, the CXCL10 knock-out improved the survival of SC-β cells in a humanized mouse model [60].
Cai et al. [61] also conducted genome-scale CRISPR screening in a mouse β-cell line (NIT-1) and found that the knock-out of Rnls, a candidate gene for type 1 diabetes identified through genome-wide association studies (GWASs), rendered the β cells resistant to autoimmune destruction. Further mechanistic investigation revealed that Rnls knock-out conferred cell resistance to ER stress and apoptosis induced by inflammatory cytokines such as IL-1β and IFN-γ. In line with these observations, Rnls overexpression had the opposite effect, promoting the autoimmune destruction of NIT-1 cells. Inspired by these results, RNLS was knocked out in human iPSCs and the edited cells were differentiated into SC-β cells. While this genetic modification did not affect the differentiation efficiency, it conferred resistance to thapsigargin-induced ER stress [61].
Several studies employed viral transduction to introduce additional genetic materials into SC-β cells due to its high efficiency in gene delivery. For instance, lentiviral delivery was employed to induce the overexpression of the immune checkpoint protein PD-L1, a strategy aimed at protecting stem cell-derived islet-like organoids from immune-mediated destruction [44]. The examples previously discussed also utilized viral vectors for the overexpression of CD47 and CD64 [54,57]. While viral transgene expression is highly efficient, it comes with potential safety concerns. These include the possibility of an immune response to viral factors and the unpredictable random insertion of DNA fragments in the cell's genome, which could lead to unforeseen adverse effects such as oncogenicity [62-64]. Alternatively, a more precise approach involves knocking in transgenes at specific genomic safe harbor loci, such as AAVS1 or CLYBL, through genome editing. This strategy offers several advantages over viral vector-mediated gene integration. It ensures that resulting cells undergo predictable and precise genomic changes [58], which can significantly facilitate the clinical translation of the engineered cell therapeutics.
Genome editing for diabetes modeling and patient-specific therapies
Basic investigations into β-cell biology have traditionally relied on the use of rodent β-cell lines due to the lack of suitable human β-cell models that accurately display mature β-cell characteristics. However, it is crucial to note that human and rodent β cells exhibit significant differences [25]. For example, human β cells possess a single insulin gene (INS), while rodent β cells have dual insulin genes (Ins1 and Ins2). Indeed, many findings derived from rodent β cells do not necessarily translate to human β cells. The rapid development in technologies for generating SC-β cells has opened the door to using them as reliable sources of human β-cell models. When combined with genome editing that can introduce any genetic variant into the genome, SC-β cells could be a superior choice for studying genetic aspects of diabetes (Fig. 3B).
For example, neonatal diabetes arising from the mutations in the INS gene can be modeled using SC-β cells edited through CRISPR/Cas9. In a study by Balboa et al. [65], patient-derived iPSCs were initially generated to study the effect of INS mutations that led to proinsulin misfolding, specifically mutations at cysteine residues critical for disulfide bond formation (C96R or C109Y). As precise investigations into the role of mutations can be conducted only in isogenic cell lines, they employed CRISPR/Cas9-based genome editing on the patient-derived iPSCs to correct the mutations via the HDR pathway. The resulting mutants and corrected iPSCs were then differentiated into SC-β cells. In-depth investigations revealed that these INS mutations induced ER stress in β cells, leading to impaired proliferation. When transplanted in vivo, the mutant cells exhibited reduced insulin secretion, increased ER stress, decreased cell size, and diminished mTOR signaling. Interestingly, however, β-cell apoptosis was not increased by these mutations [65]. This study exemplifies how genome editing technologies can be harnessed to precisely study the functional consequences of diabetes-causing mutations within an isogenic background.
Similarly, in a study by Panova et al. [66], genome editing was employed to investigate the role of a mutation located in the second intron of INS (c.188-31G>A). Patient-derived iPSCs were corrected using CRISPR/Cas9 and subsequently differentiated into SC-β cells with an isogenic background. This investigation revealed that the intronic mutation created a new splicing site, resulting in the generation of a novel splicing variant (with a 29-nt insertion compared to the wild type). Consequently, a reduction in insulin production was observed in the mutant cells [66].
Mutations in other genes can also be studied using a similar approach. For example, an iPSC line was derived from a congenital hyperinsulinism patient who carried a mutation (V187D) in the ABCC8 gene, which encodes the sulfonylurea receptor 1 (SUR1) subunit of the KATP-channel [67]. An isogenic wild-type iPSC line was then generated via CRISPR/Cas9. When these iPSCs were differentiated into SC-β cells, the SUR1-mutant cells displayed a significantly higher rate of proliferation than their wild-type counterparts. In addition, the mutant cells secreted more than threefold higher levels of insulin even under low-glucose conditions. When transplanted in vivo, these cells continued to secrete insulin even under hypoglycemic conditions, consistent with the in vitro findings. This study exemplifies how genome editing techniques facilitate the exploration of a previously unexplored role for the KATP-channel during human islet development [67].
PDX1 is a crucial transcription factor that serves as a master regulator of pancreas development and β-cell function [68]. In a study by Wang et al. [68], iPSC lines carrying known PDX1 mutations were generated using CRISPR/Cas9 technology. These mutations included homozygous P33T or C18R variants, as well as a PDX1-haploinsufficient model. Subsequently, the mutant iPSCs were then differentiated into SC-β cells. All the mutant cells displayed reduced GSIS in comparison to their wild-type counterparts. Notably, the P33T mutation had more pronounced detrimental effects on the expression of β-cell markers. Specifically, the P33T mutation downregulated genes bound by PDX1, revealing the mechanism underlying its adverse impact on β cells.
Genome editing was also employed to investigate variants of GATA6, a critical regulator during pancreas development, in patients with pancreatic agenesis (PA). To study the effects of q heterozygous 4-bp duplication in GATA6 under an isogenic background, a patient-derived iPSC line was correct to restore wild-type sequence. Conversely, a wild-type ESC line was edited to introduce the heterozygous GATA6 mutations. These 2 systems were used to explore the role of the GATA6 variants during the pancreatic differentiation toward SC-β cells. The findings revealed that GATA6 haploinsufficiency led to defects in pancreas progenitors and a shift in cell fate, as evidenced by a decrease in pancreas-specific gene expression and an upregulation of genes associated with stomach development [69] Additionally, the study identified a single-nucleotide polymorphism (SNP) located downstream of the GATA6 coding sequence in PA patients. To model this variant, CRISPR/Cas9-based knock-in was performed in ESCs. The differentiation of the resulting cells indicated that the SNP decreased GATA6 expression, negatively affecting the pancreas development and resulting in significantly reduced fraction of SC-β cells [69].
While precise HDR-based knock-in technologies were utilized in the studies mentioned earlier, NHEJ or MMEJ-based knock-out is a simpler approach and can be employed to model gene disruptions in diabetes (Fig. 1). Shi et al. [70] generated GATA6 knock-out cells using CRISPR/Cas9. This genome editing allowed the creation of both heterozygous and homozygous knock-out ESC lines. Upon differentiation of theses edited ESCs, it was observed that the loss of one GATA6 allele led to impaired generation of β-like cells. Notably, this GATA6 haploinsufficiency-related phenotype is not evident in mouse β cells, highlighting the significance of SC-β cells in modeling diabetes when combined with genome editing.
In an early study aimed to model type 2 diabetes-associated genetic variants identified through GWASs, Zeng et al. [71] used CRISPR/Cas9 to knock out the CDKAL1, KCNQ1, or KCNJ11 genes in ESCs. These specific genes were chosen since they are known to be associated with β-cell function rather than insulin resistance in peripheral tissues, making them suitable candidates for investigation using SC-β cells. The ablation of CDKAL1, KCNQ1, or KCNJ11 did not impair the differentiation process toward β-like cells, as evidenced by the expression of mature β-cell markers and the fraction of insulin-positive cells. However, all mutant cells exhibited an inability to increase insulin secretion in response to high glucose concentrations in vitro, and they failed to maintain glucose homeostasis when transplanted into streptozotocin-induced diabetic mice. Notably, CDKAL1 knock-out SC-β cells displayed hypersensitivity to glucolipotoxicity, while the knock-out of KCNQ1 or KCNJ11 did not impact cell death induced by glucolipotoxicity. A remarkable feature of genome-edited SC-β cells is their potential for developing personalized medicines targeting specific gene variants. To demonstrate this potential, the authors conducted phenotype-based high-throughput chemical screening and identified a small molecule (T5224) that selectively rescued CDKAL1-deficient SC-β cells from glucolipotoxicity. Furthermore, this compound specifically improved GSIS in these cells. As T5224 is an inhibitor of the FOS/JUN activator protein-1, they further investigated the role of the FOS/JUN pathway in β-cell dysfunction. Interestingly, the CRISPR-based knock-out of FOS was found to enhance GSIS in vivo [71].
Cardenas-Diaz also harnessed CRISPR/Cas9-based knock-out to study the most common monogenic form of diabetes arising from mutations in HNF1A [72]. In this study, heterozygous or homozygous HNF1A knock-out ESCs were differentiated into SC-β cells. The analysis of endocrine hormone expression patterns revealed a decrease in insulin-positive cells and an increase in glucagon-positive cells among the stem cell-derived cells. In-depth investigation revealed that HNF1A deficiency drove endocrine differentiation toward a more alpha cell-like gene expression signature. Furthermore, the study demonstrated that HNF1A is necessary for optimal insulin secretion in SC-β cells. These findings offer a perspective distinct from observations in rodent models, emphasizing the significance of genome editing and SC-β cells in advancing diabetes research [72].
GWASs have been extensively conducted to identify risk factors associated with type 1 diabetes and type 2 diabetes. Since type 1 diabetes results from β-cell failure, modeling this condition using SC-β cells has emerged as an essential tool for in-depth research. Intriguingly, many genetic variants associated with type 2 diabetes are linked to β-cell development or function [73], highlighting the need for investigations utilizing SC-β cells in conjunction with genome editing.
Patient-derived iPSCs represent invaluable research assets for studying the mechanisms of genetic diabetes as discussed earlier. Beyond mechanistic investigations, these cells hold great promise for advancing the development of patient-specific autologous cell-based therapeutics for diabetes. Particularly, single pathogenic variants can be corrected by one-step genome editing, making then an ideal starting point for the creation of patient-specific cell therapies.
To demonstrate the feasibility of autologous cell therapies for neonatal diabetes caused by INS mutations, Ma et al. [74] obtained iPSCs from a neonatal diabetes patient who had an INS mutation at the translation start site (ATG to ATA). Using a CRISPR/Cas9-based knock-in approach, they successfully restored the start site to produce corrected iPSCs. Subsequently, these corrected iPSCs were differentiated into SC-β cells. While the uncorrected iPSCs could be converted into endocrine cells, as evidenced by the expression of several endocrine markers, they failed to produce insulin. In contrast, the corrected iPSCs were capable of differentiating into insulin-producing β-like cells. Remarkably, those cells exhibited the ability to restore normal glucose homeostasis in vivo, including the precise regulation of insulin secretion in response to metabolic changes [74]. Even though there were various issues encountered in this study, such as teratoma formation from transplants and off-target effects of genome editing, the value of the gene-correction strategy was clearly demonstrated, highlighting its potential for use in autologous cell therapies.
Maxwell et al. [75] derived iPSC lines from fibroblasts of individuals with Wolfram syndrome (WS). WS is an autosomal recessive disorder caused by variants in the WFS1 gene, resulting in ER stress, unfolded protein response, and subsequent β-cell death. Since there is currently no standard therapy for WS or ER stress-related disease, the authors aimed to generate gene-corrected, patient-derived SC-β cells. First, a CRISPR/Cas9-based knock-in approach in iPSCs was employed to correct the pathogenic variants. Subsequently, these edited cells were differentiated into SC-β cells. Remarkably, the corrected SC-β cells exhibited significantly increased insulin secretion coupled with an appropriate response to glucose challenges. Consistent with these findings, there was a significant increase in the fraction of cells displaying β-cell markers. Importantly, when transplanted into mice with preexisting diabetes, the corrected SC-β cells successfully restored normoglycemia. Mechanistic investigations into the corrected SC-β cells revealed that the relief of ER stress was attributed to the gene correction [75].
Autologous cell therapies hold significant promise as they do not require the administration of immunosuppressants. This approach is particularly attractive for diabetes caused by single mutations, which account for approximately 1% to 5% of all diabetes cases [74]. Correcting these monogenic variants through genome editing offers a straightforward solution to treat the disease. For other types of diabetes arising from mutations in multiple genes, the rapid advancement of genome editing technologies would enable multiplex genome editing. This approach would allow the simultaneous correction of multiple genes, thereby enhancing the efficiency and potentially reducing the cost of autologous cell therapies.
Genome editing in primary pancreatic islets
Genome editing has been primarily performed in PSCs due to their amenability to editing, rapid proliferation, and the ability to develop into clones. The edited PSCs are then differentiated into gene-corrected β-like cells. While these SC-β cells hold promise as an abundant source for therapeutic development, primary pancreatic islets have historically been used for developing cell-based therapeutics for diabetes because of their mature β-cell function. Indeed, a primary islet-derived cell therapy was recently approved by the Food and Drug Administration for treating diabetes [9,10]. However, the use of primary islets present challenges, such as the need for immunosuppressants administration to prevent allograft rejection. Thanks to rapid advances in CRISPR technologies, genome editing can now be applied to primary islets, addressing several issues associated with current islet-based cell therapies.
Bevacqua et al. [76] demonstrated the feasibility of CRISPR/Cas9-based knock-out in primary islets. The study began by successfully disrupting 2 critical genes for β-cell functions, PDX1 and KIR6.2. Indeed, the knock-out impaired β-cell regulation and function. To further validate their approach, the researchers explored the role of non-coding DNA sequences through genome editing. Specifically, they identified a diabetes risk SNP located at the KCNJ11-ABCC8 or SIX2-SIX3 non-coding locus. Then, they introduced small deletions at the SNP locus by delivering Cas9 along with 2 gRNAs. This editing impaired β-cell function, revealing the significance of non-coding regulatory elements in type 2 diabetes [76].
For developing primary islet-derived cell therapeutics, hypoimmune engineering was performed in primary islets, so that the transplant could survive without immunosuppression. Hu et al. [54,77] employed CRISPR/Cas9 to knock out B2M and CIITA, while simultaneously overexpressing CD47 through viral delivery. The resulting hypoimmune islets survived and ameliorated diabetes in immunocompetent allogeneic humanized mice. Moreover, the engineered islets could be protected from autoimmune destruction in autologous, diabetic humanized mice.
One of the major limitations in genome editing of primary islets is that the editing method is restricted to knock-out. HDR-based knock-in is not feasible in primary islets because the HDR pathway is active only in dividing cells, but not in primary β cells that usually do not proliferate. Nevertheless, CRISPR-based knock-out coupled with transgene delivery through viral vectors could offer a viable approach to broaden the application of gene-edited primary islets in therapeutics.
Limitations of CRISPR/Cas9-based genome editing and potential solutions
While genome editing is an extraordinary tool for creating designer cell therapeutics, there always exist concerns about off-target gene editing [36,78-80]. Unintended genomic changes at non-targets can potentially affect cell fitness leading to cell death. The cells may be converted into non-functional cells or even into cancerous cells. As such, precautions must be taken to avoid off-target editing risks. For example, bioinformatics tools should be employed to design gRNAs with minimal off-target potential. In cases where only a limited set of gRNAs can be used, and the chances of off-target effects are high (e.g., when correcting a specific gene variant through HDR), recently developed anti-CRISPR tools may be employed to mitigate off-target effects [36,81]. Whenever possible, whole-genome sequencing of edited stem cell clones should be conducted to assess the potential for off-target modifications. If whole-genome sequencing is unavailable, it is advisable to examine several high-confidence off-target loci to ensure the safety of the cell therapeutics.
The genome-editing examples mentioned above primarily employed CRISPR/Cas9-mediated knock-out or donor DNA-assisted knock-in techniques (Fig. 1). In addition to these conventional tools, emerging genome editing methods, such as base editors or prime editors, could be employed for more precise gene editing. Base editors can introduce single-base exchanges, while prime editors can make small insertions, deletions, and base exchanges without causing DNA DSBs, unlike Cas9 [82]. Consequently, base editors and prime editors display significantly reduced off-target effects, making genome editing safer. Although their editing efficiencies may not match that of Cas9, rapid advancements in genome editing technologies are paving the way for their widespread application in PSCs, particularly for introducing small genomic changes (e.g., modeling disease-related gene variants and correcting disease-causing variants).
CRISPR/Cas9-mediated HDR should be employed when introducing large gene fragments into PSCs (Fig. 2). However, HDR efficiency is typically low, necessitating the use of additional techniques for generating gene-edited cells. These techniques include the use of small-molecule inhibitors of the NHEJ pathway, which competes with the HDR pathway [83], chemical modification of donor DNA [84], and conjugating donor DNA to Cas9 [62], among others. A combination of multiple methods can be employed to achieve maximal HDR efficiency. Nonetheless, it is important to exercise caution when applying these techniques. For example, small-molecule inhibitors of the NHEJ pathway could have detrimental effects on certain cell types, as NHEJ is a constitutively active pathway that is important for cellular fitness.
Alternatively, methods for isolating knock-in clones can be employed. Genes for antibiotic resistance proteins or fluorescent proteins are commonly introduced into the genome along with the desired knock-in fragment to facilitate the isolation of gene-edited PSCs. However, the introduction of these reporter genes is inevitably accompanied by the introduction of additional regulatory factors (e.g., promoters, terminators, or polycistronic expression cassette). Since these elements often originate from viral sequences, it is essential to consider whether the new sequences in the genome might provoke adverse effects in vivo [63,64].
Regardless of the type of editing, undesirable genomic changes at the on-target site should also be investigated. These on-target effects may involve large deletions or insertions at one allele, while the other allele carries the correct edits. As these effects cannot be detected through standard Sanger sequencing, it is recommended to perform quantitative polymerase chain reaction-based copy number quantification when isolating correct PSC clones [85]. Another on-target effect to consider is the loss of heterozygosity, which can be identified by genotyping a neighboring SNP [85]. These approaches can ensure the safe use of gene-edited PSCs.
Future perspectives
With rapid advancements in genome editing, researchers can now precisely modify therapeutic cells to confer them with new functionalities. The most notable achievements in this field include enhancing GSIS and developing strategies for evading immune destruction. Since there still exist diverse issues to be solved for the practical application of β-cell therapeutics, genome editing is poised to play an even more prominent role in future research.
For example, β-cell therapeutics often face challenges related to hypoxia and subsequent cell death due to inadequate vascularization to support the survival of transplants, especially in the early stages after transplantation [17]. Genome editing can be employed to enhance the resistance of these cells to hypoxia-induced apoptosis, maximizing their therapeutic potential. Recent clinical trials have utilized macroencapsulation devices for protecting SC-β cells from immune attacks. However, foreign body responses against the transplanted device have led to device fibrosis and failure [11,13]. Genome editing could also offer a solution to this issue, for instance, by engineering cells to locally secrete anti-fibrosis factors.
Nonetheless, transplanted cell therapies carry inherent safety risks, as stem cell-derived cells may potentially develop into teratomas [21,74]. Therefore, it is crucial to develop safety switches for selectively eliminating transplants when adverse effects arise. The need for safety switches becomes even more apparent when transplants are engineered to be hypoimmunogenic, as these cells cannot be naturally eliminated by the patient's immune system even in the presence of adverse effects. Hence, there is a critical requirement for precise control over these therapeutic modalities. In the conventional Edmonton protocol and recent clinical trials, primary islets or SC-β cells are transplanted directly into the portal vein, in combination with immunosuppressive drug administration, without encapsulation [9,10]. This direct infusion of cells necessitates additional safeguards to mitigate potential adverse effects.
These requirements can be addressed through genome editing techniques designed to incorporate safety switches into cell therapeutics. For instance, the CRISPR/Cas9-based gene knock-in method was employed to introduce a drug-inducible caspase-9 (iCasp9) safety switch into PSCs [86]. This switch was integrated at the C-terminus of NANOG, along with a 2A self-cleaving peptide [87]. Thus, the safety switch is specifically expressed in PSCs since NANOG is a stem cell-specific transcription factor not expressed in differentiated cells. Upon treatment with a specific small molecule, the caspase-9 protein dimerizes and induces the selective death of undifferentiated stem cells, effectively addressing concerns related to teratoma formation [87]. When the iCasp9 switch is knocked in at the C-terminus of ACTB, a housekeeping gene expressed in all cell types, both undifferentiated and differentiated cells were eliminated upon treatment with a small molecule. Notably, all these switches efficiently worked in mouse models to remove the transplanted cells when the corresponding small molecules were administered [87]. Similar genome-editing strategies could be employed in β-cell therapeutics to ensure their safe use in future research.
In summary, SC-β cells can play crucial roles as sources of cell-based therapeutics for diabetes. They also serve as valuable models for human β cells, as obtaining mature human β-cell lines is challenging. When combined with genome editing techniques, SC-β cells will offer a powerful platform for both treating diabetes and conducting in-depth studies on the genetic aspects of diabetes.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print